
 buy artwork
buy artwork
 buy artwork
buy artwork
π Day 2025 Art Posters - TTCAGT: a sequence of digits















On March 14th celebrate `\pi` Day. Hug `\pi`—find a way to do it.
For those who favour `\tau=2\pi` will have to postpone celebrations until July 26th. That's what you get for thinking that `\pi` is wrong. I sympathize with this position and have `\tau` day art too!
If you're not into details, you may opt to party on July 22nd, which is `\pi` approximation day (`\pi` ≈ 22/7). It's 20% more accurate that the official `\pi` day!
Finally, if you believe that `\pi = 3`, you should read why `\pi` is not equal to 3.

Well—well; the sad minutes are moving,
Though loaded with trouble and pain;
And some time the loved and the loving
Shall meet on the mountains again!
—Emily Bronte
Welcome to this year's celebration of `\pi` and mathematics.
The theme this year is Sanger sequencing — old-school, one base at a time.
This year's `\pi` poem is Loud Without The Wind Was Roaring by Emily Bronte.
This year's `\pi` day song is Movements by Luca Musto.
Also, the tabbed menu above is full. Gasp.



I work in a genome center, so it was just a matter of time before one of the Pi Day celebrations encoded the digits of `\pi` as a sequence of nucleotides (A, T, G and C). It took 12 years.
Our first sequencer was the MegaBACE 1000, which now exists only in old photos).
Unfortunately, there was nothing "mega" about it. You were lucky to sequence 500 contiguous bases of DNA. So, halfakiloBACE?
Under the hood, the sequencing was done with the Sanger method.
Check out the method section to learn how Sanger sequencing works.
But briefly, it's a sequencing method in which bases are identified from a series of fluorescence peaks. This is why the art has peaks!


The posters show `\pi` up to the Feynman Point, which are six 9's at decimal places 762–767.

Each digit is encoded by two bases:
0 GA 1 CA 2 TC 3 TT 4 GT 5 GC 6 AA 7 CC 8 TA 9 GG
With this scheme, 3.14 reads as TTCAGT. Hence, the title of the art “TTCAGT: a sequence of digits.”

Explore the art posters.
 buy artwork
buy artwork
Propensity score weighting
It is not certain that everything is uncertain. —Blaise Pascal
We have already explored how we can mitigate bias caused by confounding variables in observational studies using propensity score (PS) matching (PSM) and propensity score weighting (PSW). However, any statistical model is only as good as its assumptions and, if it is specified incorrectly, it can itself produce biased estimates of the treatment effect.
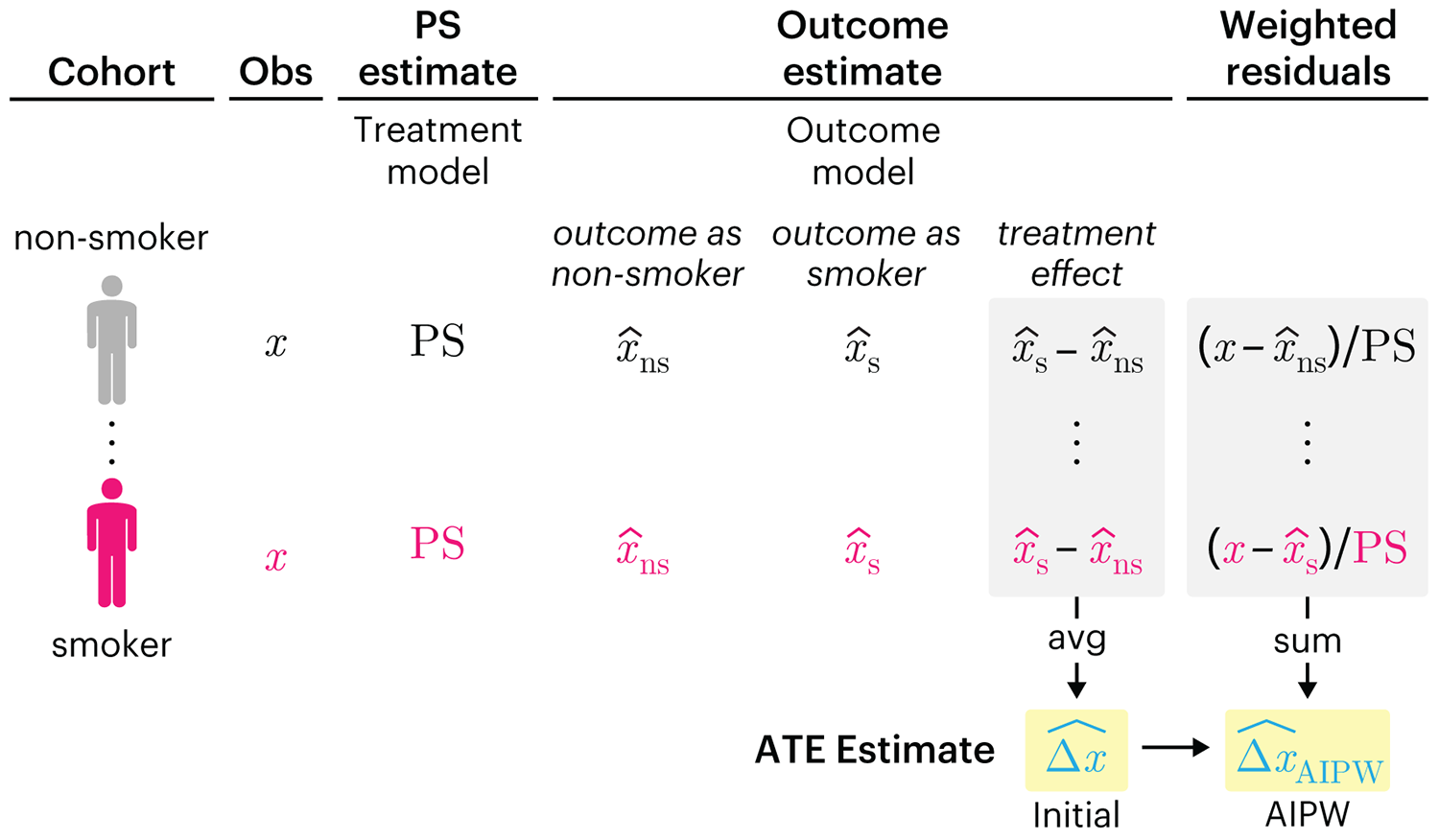
This month, we explore double robustness, a powerful statistical concept that provides a valuable “safety net” against the risk of an incorrect model. It offers two opportunities, instead of just one, to obtain a valid estimate of the treatment effect — making it possible to draw credible causal inferences from observational data without having to depend on a single set of modeling assumptions.
Kurz, C.F., Krzywinski, M. & Altman, N. (2026) Points of significance: Double Robustness. Nat. Methods 23:868–869.
Nature Biotechnology cover
My cover design on the 7 April 2026 Nature Biotechnology issue shows the dendrogram that represents a cluster of uniquely expressed (or downregulated) genes in human naive stem cells induced from such cells. Within each dendrogram block, the genomic barcode sequence (sampled from Supplementary Table 1) is depicted with a Code 39 barcode. The highlighted barcode is one of those used for cell isolation.
Ishiguro S. et al. A multi-kingdom genetic barcoding system for precise clone isolation (2026) Nature Biotechnology 44:616–629.

Browse my gallery of cover designs.

Happy 2026 π Day—
Art for the 5%
Celebrate π Day (March 14th) and enjoy the art — but only if you're part of the 5%.
Go ahead, see what you can't see.
Ishihara's Tests for Colour Deficiency
Authentic and accurate images of Ishihara's test plates photographed (and lovingly color-corrected) from the 38-plate Ishihara's Tests for Colour Deficiency.
I also provide the position, size, and color of each circle on each test plate.


Symmetric alternatives to the ordinary least squares regression
What immortal hand or eye, could frame thy fearful symmetry? — William Blake, "The Tyger"
This month, we look at symmetric regression, which, unlike simple linear regression, it is reversible — remaining unaltered when the variables are swapped.
Simple linear regression can summarize the linear relationship between two variables `X` and `Y` — for example, when `Y` is considered the response (dependent) and `X` the predictor (independent) variable.
However, there are times when we are not interested (or able) to distinguish between dependent and independent variables — either because they have the same importance or the same role. This is where symmetric regression can help.

Luca Greco, George Luta, Martin Krzywinski & Naomi Altman (2025) Points of significance: Symmetric alternatives to the ordinary least squares regression. Nat. Methods 22:1610–1612.
Beyond Belief Campaign BRCA Art
Fuelled by philanthropy, findings into the workings of BRCA1 and BRCA2 genes have led to groundbreaking research and lifesaving innovations to care for families facing cancer.
This set of 100 one-of-a-kind prints explore the structure of these genes. Each artwork is unique — if you put them all together, you get the full sequence of the BRCA1 and BRCA2 proteins.
