


DNA on 10th — Science this way
contents
Design is an exploration with no ultimate destination—just snapshots of views along the way. The methods and process behind the active process of design—as well as the many tangents not taken—are as interesting (if not more) than the final product, which is static.
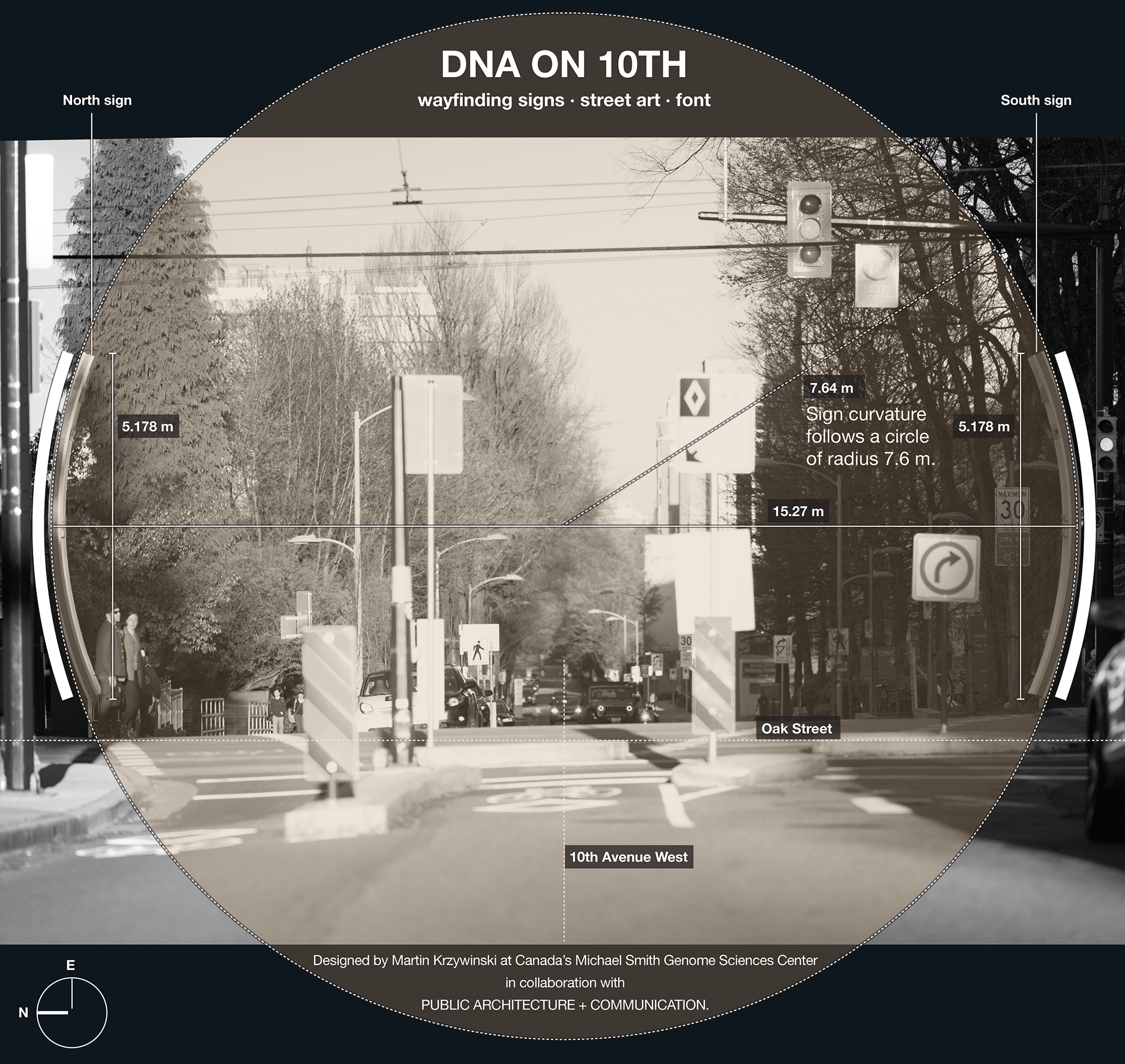
The design of the signs is motivated by the research and patient care that takes place everyday in the buildings along the 10th Avenue Health Care Corridor.
For most of my projects, I create a detailed writeup about their design.




We turn our attention to genomes—the way astronomers turn their attention to the skyes—to understand disease. Just like telescopes take many images over a long period of time, so do our sequencers, which are chemical photostudios that collect and analyze millions of individual images over several days of a "sequencing run".
These machines are our workhorses of data collection and the beginning of new understandings. At Canada's Michael Smith Genome Sciences Centre, which is just a couple of blocks east of the signs, we have sequenced about 2.3 petabases (I just checked [29 Mar 2019, 10:09am PDT] and the actual number of 2,288,999,145,239,301).
It seemed fitting to feature some of these bases on the signs. Even though the signs are just over 5 m tall and generously allot 3,875 mm × 851 mm for the art, there wasn't enough room for all 2.3 petabases, so I picked 4,203 that I felt were important (2,105 on the south sign and 2,098 on the north sign).
Just as well, though. Showing everything is dull—the value is always in the selection. “To show something, hide something.” is what they say in Japan.
There's plenty of room at the bottom. —Richard Feynman
If we wanted to show all the bases on the sign, how big would each base have to be to fit them all in? Let's approximate this. $$\sqrt{\frac{3.878 \times 0.851}{2\,288\,999\,145\,239\,301}} = 3.8 \times 10^{-8} \text{m} = 38 \text{ nm}$$
Each square millimeter would have to hold about 700 million bases and each base would have to fit in a 38 nm × 38 nm square. 38 nm is the length of about 11 twists of the DNA double helix (or 110 bases, see below). Given that the DNA double helix is 2 nm is diameter, we could place 18 pieces of DNA of 11 twists each within this square, or about 2,000 bases. I'm neglecting the fact that this would technically be 2,000 base pairs.
So if we wanted to encode all our bases on the sign, we could accept an encoding system that is 2,000 less efficient than DNA.
However, by the time we have sequenced about `6 \times 10^{18}` bases (6 exabases), we would no longer be able to make this compromise—this is how many bases would fit on the sign if stored in their native double helix format, with the DNA molecules side-by-side.
The orientation, color and brightness tells the story of mutations and disease. The signs are backlit and best viewed in the evening or at night. The signs' art is based on a 18 × 93 grid. At each point, a shape roughly the size (30 mm) of a twonie (the Canadian two dollar coin which is 28 mm in diameter) represents bases in the human genome.

As you may know, DNA is a double helix—but not just any double helix.
A variety of helices are possible :A-DNA, B-DNA, Z-DNA to name a few. These vary in the tightness and direction of winding.
The DNA in our cells is B-DNA. It is right-handed—if you hold out your right hand and make a first the the thumb pointing up, the helix will wind in the direction of your curled fingers.
A full turn of B-DNA is about 34 Å (3.4 nm) along its axis. Distance between bases along the axis is about 3.4 Å—each turn is 10 bases. I made the design choice to stretch the helix on the sign vertically because the sign is so much taller than it is wide. On the sign, each turn has about 24 bases.
Its ratio of the size of the minor groove to the major groove is about 13:21 and this defines the offset between the two strands when they are drawn in two dimensions. This groove ratio is accurately depicted on the sign.
The helix is confined within the center 16 columns of the sign and because the sign shapes are arranged on a grid, takes on a flat shape towards the edges of the sign.
In the phosphate-sugar backbone of DNA, the phosphate group is attached to the 5' carbon, which is considered to be the "upstream" position. The hydroxyl group is attached to the 3' carbon, which is the "downstream" position. The two backbones have opposite orientation—one runs 5'→3' and the other 3'→5'. Nucleic acids are always synthesized in the 5'→3' direction.
The strand that starts first in the bottom of the sign is the 5' strand—this is an arbitrary choice, but one that determines subsequent design elements. This strand is shown in red in the helices on the right.
Let each grid point coordinate be given by `(x,y)` where `x` is the column (from the left) and `y` is the row (out of `N` rows, from the bottom). Let `k=1` for the 5' strand and `k=0` for the 3' strand. $$ x = 1 + \Bigl\lfloor{\tfrac{1}{2}\left[ 1 + \text{sin}\left( 2 \pi \left( \tfrac{2(y+10)}{N} + \tfrac{13k}{34} \right) \right) \right) }\Bigr\rceil $$
The z-depth of the strand has the same equation except now `\text{sin}(\cdot)` is `\text{cos}(\cdot)`.
These parameters (general DNA geometry equations) combine the offset of the helix, offset of the 5' strand (to approximate the minor-to-major groove ratio) and accommodate 2 periods of the helix on the sign.
Because the position of the helix snaps to the closest grid point, in some parts the helix appears jagged. I fiddled with the helix offset to limit the impact of this.
I selected the helix offset to have the two strands cross roughly at the height of the average viewer.
There is a geometrical relationship between how the helix is depicted on the south and north signs.
The north sign shows the helix as it would appear if you were looking at it from behind the south sign. Thus, just as on the south sign the 5' strand is the one that initially receedes away from the viewer, on the north sign the 5' strand comes towards the viewer (and the 3' receedes).
It would be a mistake to simply mirror the images, since that would change the orientation from right-handed to left-handed.
The next step is to lay down some sequence along the helix. But which sequence?
In the context of cancer, there is no gene more famous than the "guardian of the genome", the TP53 gene. This gene codes for the tumor suppressing protein p53, which plays a vital role in the process of apoptosis—cell self-destruct if they cannot repair their DNA. Mutations in this gene create malfunctioning p53 protein which limit the effectiveness of this protection.
In our Personal Oncogenomics Program we have sequenced many patients with mutations in TP53. Here are eight such mutations (of many):
chr:pos snp protein mutation sequence
17:7577534 C>A Arg249Ser GTGATGATGGTGAGGATGGG[C/A]CTCCGGTTCATGCCGCCCAT
17:7577538 C>T Arg248Gln TGATGGTGAGGATGGGCCTC[C/T]GGTTCATGCCGCCCATGCAG
17:7577539 G>A Arg248Trp GATGGTGAGGATGGGCCTCC[G/A]GTTCATGCCGCCCATGCAGG
17:7577547 C>A Gly245Val GGATGGGCCTCCGGTTCATG[C/A]CGCCCATGCAGGAACTGTTA
17:7577547 C>T Gly245Asp GGATGGGCCTCCGGTTCATG[C/T]CGCCCATGCAGGAACTGTTA
17:7577556 C>T Cys242Tyr TCCGGTTCATGCCGCCCATG[C/T]AGGAACTGTTACACATGTAG
17:7577559 G>A Ser241Phe GGTTCATGCCGCCCATGCAG[G/A]AACTGTTACACATGTAGTTG
17:7577568 C>T Cys238Tyr CGCCCATGCAGGAACTGTTA[C/T]ACATGTAGTTGTAGTGGATG
As you can see, all these occur within a short span of the gene. I've composited the reference sequence and mutations below
7577534 GTGATGATGGTGAGGATGGG[C/A]CTCC G GTTCATGC C GCCCAT 7577538 TGATGGTGAGGATGGGC CTC[C/T]G GTTCATGC C GCCCATGC AG 7577539 GATGGTGAGGATGGGC CTCC [G/A]GTTCATGC C GCCCATGC AGG 7577547 GGATGGGC CTCC G GTTCATG[C/A]C GCCCATGC AGG AACTGTTA 7577547 GGATGGGC CTCC G GTTCATG[C/T]C GCCCATGC AGG AACTGTTA 7577548 GATGGGC CTCC G GTTCATGC [C/T]GCCCATGC AGG AACTGTTAC 7577556 TCC G GTTCATGC C GCCCATG[C/T]AGG AACTGTTAC ACATGTAG 7577559 G GTTCATGC C GCCCATGC AG[G/A]AACTGTTAC ACATGTAGTTG 7577568 C GCCCATGC AGG AACTGTTA[C/T]ACATGTAGTTGTAGTGGATG
which can be combined into a single view of the reference and mutations at each location
0 10 20 30 40
01234567890123456789012345678901234567890123456
TGATGATGGTGAGGATGGGaCTCtaGTTCATGatGCCCATGtAGaAACTGTTAtACATGTAGTTGTAGTGGATG
......*...**.......**.......*..*........*.....
reference C CG CC C G C
mutations A TA AT T A T <- mutation used
T <- mutation not used
The sequence along the 5' helix is given by the mutated 47-base TP53 gene sequence in red above: GATG...ATGT, where the mutated bases are shown in lowercase. For positions in which more than one mutation has been observed across patients, I've arbitrarily picked one of them. The sequence runs from the bottom of the sign to the top.
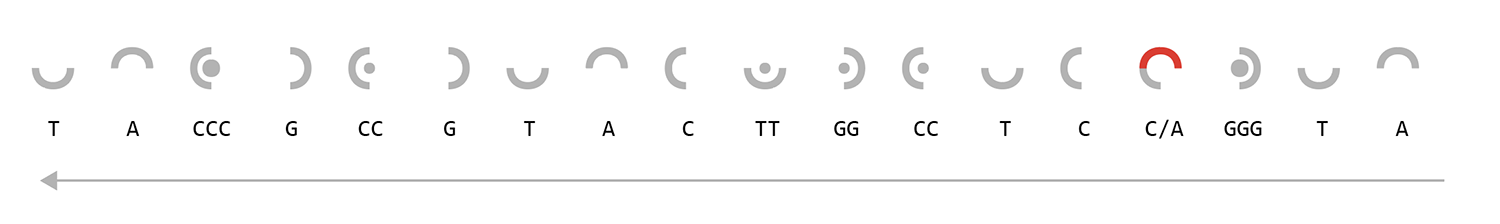
The sequence is encoded using the following scheme.

A/T) and (C/G) are mirror images of each other. Repeats are shown with a small circle (base repeats twice) or a large circle (base repeats three times). Longer repeats are shown by combinations of these shapes, as shown by the example ATTCGGGGGT sequence on the right.


The images on the left show the encoding of the mutated TP53 sequence on the 5' (red) and 3' (grey) strands. The sequence along the 3' helix is the complement of this—where A ↔ T and C ↔ G.
We have 93 rows on the sign with the 5' helix occupying 47/92 and the 3' helix the other 46/92.
From bottom to top, you can read the sequence off as
5' GATGGGaCTCtaGTTCATGatGCCCATGtAGaAACTGTTAtACATGT ||||||*|||**|||||||**|||||||*||*||||||||*||||| 3' CTACCCtGAGatCAAGTACtaCGGGTACaTCtTTGACAATaTGTAC


The TP53 sequence has mutations only at 8 positions positions, indicated by * below.
......*...**.......**.......*..*........*.....
5' GATGGGaCTCtaGTTCATGatGCCCATGtAGaAACTGTTAtACATGT
C CG CC C G C
For example, in the first position (7th base, 13th row from the bottom) the mutation is A and the reference is C.
To mark these positions, the reference sequence of TP53 runs along the corresponding rows in which the TP53 sequence along the helix has a mutation, as shown in the images on the left. And here's where things get a little fun and a little tricky. Let's look at the row on the south sign that aligns with the first TP53 mutation.
The sequence of this row is
TACCCGCCGTACTTGGCCTCCGGGTA
and it looks like this
This sequence is part of a reversed reference TP53 sequence and the way that it aligns with the mutated A is the keystone of the design.
Notice how the reference sequence has a C, here drawn in grey, at the same position where the helix sequence has an A, here drawn in red. You can use this intersection to read out the reference base and mutated base of TP53 and reconstruct (reading right-to-left) a part of the TP53 sequence
AATGGG[C/A]CTCtaGTTCATGatGCCCATGtAGaAACTGTTAtACATGT
The sequence is reversed because all rows that overlap with a mutation on the 5' strand have sequence running in the direction pointed to by the sign. This is a design decision to give both the signs and their content meaningful orientation to the way they were pointing, instead of simply running the sequence left-to-right, as we would normally read it.
Note that the sequence is only reversed for these rows but not reverse complemented. I skipped the complementing step because in the rows because I wanted to limit complementarity to the TP53 sequence along the 5' and 3' strands—these are the two sequences that are complemented in nature, whereas the rows are merely background.
The other 7 rows that intersect with the mutated TP53 bases along the 5' helix are handled in the same way. Each row samples a part of the TP53 sequence accordingly so that the mutated base along the helix overlaps with the correct reference base.
This alignment means that you can read off the TP53 sequence from the mutated position along the row (working backwards left to right) or along the helix (working backwards downward). What
This last part is one of the hidden puzzles. What makes this particular aspect challenging to spot is that any repeated bases that do not intersect with the helix can be encoded using a single symbol, whereas all the bases along the helix are explicitly encoded with the singleton symbol. Thus, as you read, for example, GGG along the helix you'll have to look for the corresponding triplet G symbol in the row.


By comparing the TP53 rows on the north and south signs, you can see that they are reversed to one another—this is because the 5' strand row sequences run in the direction pointed to by the sign.
The rest of the rows are filled with sequence from other genes in which our sequencing has observed mutations.
Just like for the TP53 rows described above, the gene sequence in these rows is its reference sequence for the gene and the sequence aligns with the helix at the position where the gene has a mutation observed.
Sequences that intersect with the 3' helix run in the opposite direction to the sign. Thus, on the south sign which points to the left, the 3' helix gene sequences run left to right.
Previously I mentioned how I selected which strand is the 5' and which strand is the 3' arbitrarily. While the choice is arbitrary, the design marks the 5' strand as special: only the rows that intersect the 5' strand's TP53 mutatations are ones that use sequence from TP53. All other rows (non-mutated TP53 5' helix bases and all complementary TP53 bases on 3' strand) sample other genes.
That's another puzzle in the sign.
I had sequence data for over 500 genes. For each row, I was constrained to select a gene whose mutation was the base of the helix. However, because there are only four possible bases, many genes matched this criteria for any row.
I originally selected the genes to sample as many different genes as I could. At the time, it seemed like a nice optimization. But then I had the idea of embedding a deeper message in the sign.
If you look at the first letters of the genes for the 93 rows on the south sign, you'll find the following text
BCCANCERCMEITCHAELSPTMTIBTHGENKZOMESCITETNCESCEKNETRE ... ... PERTSONAPTLONCOZKGEHNOMICSPTROGRAMCCNBMK
Similarly, for the north sign
BCCANCERCMRITCHAELSPTMTIFTHGENCKOMESCITETNCESCEKNLTRE ... ... PERTSONAPTLONCOTKGESNOMICSPTROGRAMCCNBMK
And if you look very closely, you'll see the following embedded in the string of letters.
BC CANCER MICHAEL SMITH GENOME SCIENCES CENTRE
PERSONAL ONCOGENOMICS PROGRAM
There may be more puzzles in the signs.
I'll leave you to figure it out.