






visualization + design
Triploid Genome Coverage Tables
Given a location `x` defined in the context of `h` chromosomes, the probability that position `x` is covered at least `\phi` times is `P_{h,\phi}` and given by $$ P_{h,\phi} = \left( 1 - \sum \frac{1}{k!} \left( \frac{\rho}{h}^k \right) e^{-\rho/h} \right)^h \tag{1} $$
For more details, see Wendl, M.C. and R.K. Wilson. 2008. Aspects of coverage in medical DNA sequencing. BMC Bioinformatics 9: 239.
For a given sequencing redundancy `\rho` (e.g. `\rho`-fold, as determined by the length of the haploid genome) of a triploid genome, the fraction of the triploid genome represented by at least `\phi` reads is reported by `P_{h,\phi}`. Coverage by fewer than `\phi` reads is reported as `1-P_{h,\phi}`. Coverage by exactly `\phi` reads is `P_{h,\phi} - P_{h,\phi+1}`. Entries for which fractional coverage is `\lt 10^{-5}` are not shown.
A rudimentary Monte Carlo simulation of genome coverage is also available, and is a useful supplement to the exact probabilities shown here.
http://mkweb.bcgsc.ca/coverage/?aneuploidy=12&depth=200
EXAMPLE 1
Suppose you carried out 3-fold redundant (`\rho=3`) sequencing of a haploid genome (`h=1`). 95.02% of the genome will be covered by at least one read (`P_{1,1}`) while 22.40% will be covered by exactly 3 reads (`P_{1,3} - P_{1,4}`).
EXAMPLE 2
You are sequencing a sample with a tumor content of 25% and you're interested in the depth of sequencing required to detect heterozygous mutations in the tumor. This scenario is equivalent to an aneuploidy = 8 genome—any given allele is present 8 times. If you sequence at (`\rho=200`), then 95% of the bases will be covered at a depth of at least `\phi = 14` (`P_{8,14} = 0.9494`). If you're satisfied with `\phi = 5` then you only need `\rho = 100` since now `P_{8,5} = 0.9580`.
ANALYTICAL vs STOCHASTIC
View plot that compares analytical vs stochastic results.
{kind=link}
HAPLOID vs DIPLOID
View plot that compares 100x and 200x coverage of haploid and diploid genomes.
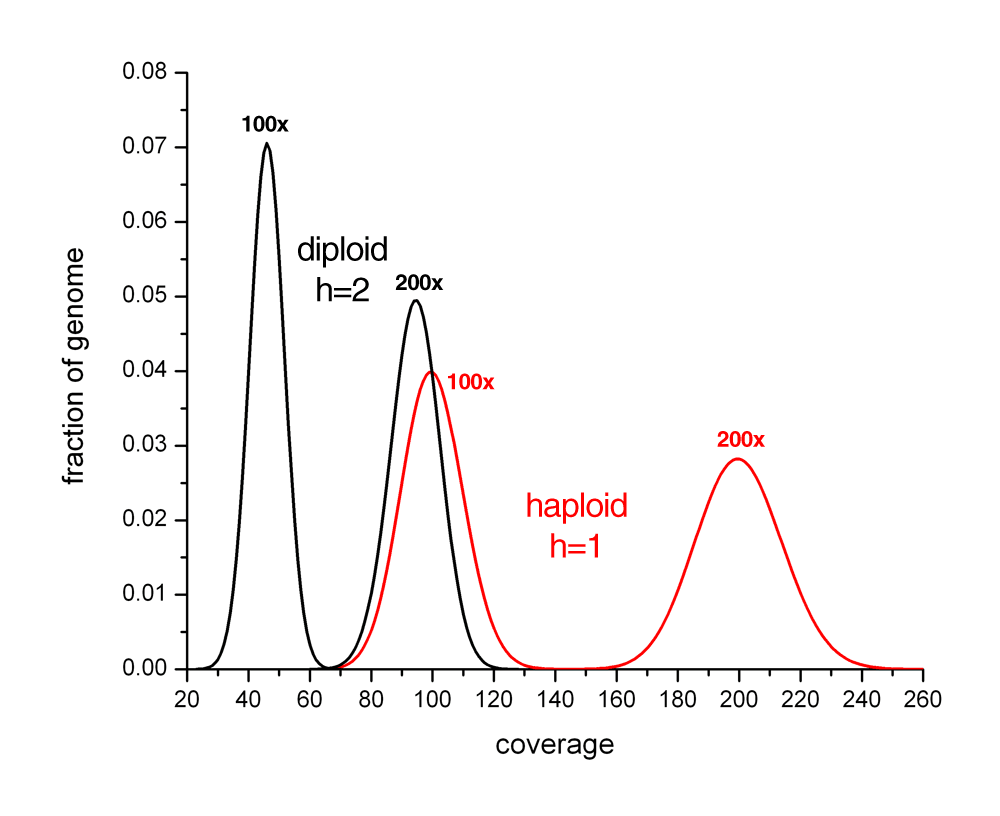{kind=link}
CODE
Download Perl scripts for analytical (to produce the tables below for any `\rho`) and stochastic coverage calculations.
sequencing redundancy for a triploid genome
View table for sequencing redundancy `\rho` = 1 2 3 4 5 6 7 8 9 10 20 25 50 75 100 of a triploid genome.
sequencing redundancy 1-fold (`\rho / h = 0.3`)
| `\phi` | `P_{h,\phi} - P_{h,\phi+1}` | `1-P_{h,\phi}` | `P_{h,\phi}` |
|---|---|---|---|
| 0 | 0.9772 | 0.0000 | 1.0000 |
| 1 | 0.0227 | 0.9772 | 0.0228 |
| 2 | 0.0001 | 0.9999 | 0.0001 |
sequencing redundancy 2-fold (`\rho / h = 0.7`)
| `\phi` | `P_{h,\phi} - P_{h,\phi+1}` | `1-P_{h,\phi}` | `P_{h,\phi}` |
|---|---|---|---|
| 0 | 0.8848 | 0.0000 | 1.0000 |
| 1 | 0.1122 | 0.8848 | 0.1152 |
| 2 | 0.0030 | 0.9970 | 0.0030 |
| 3 | 0.0000 | 1.0000 | 0.0000 |
sequencing redundancy 3-fold (`\rho / h = 1.0`)
| `\phi` | `P_{h,\phi} - P_{h,\phi+1}` | `1-P_{h,\phi}` | `P_{h,\phi}` |
|---|---|---|---|
| 0 | 0.7474 | 0.0000 | 1.0000 |
| 1 | 0.2341 | 0.7474 | 0.2526 |
| 2 | 0.0179 | 0.9815 | 0.0185 |
| 3 | 0.0005 | 0.9995 | 0.0005 |
sequencing redundancy 4-fold (`\rho / h = 1.3`)
| `\phi` | `P_{h,\phi} - P_{h,\phi+1}` | `1-P_{h,\phi}` | `P_{h,\phi}` |
|---|---|---|---|
| 0 | 0.6007 | 0.0000 | 1.0000 |
| 1 | 0.3423 | 0.6007 | 0.3993 |
| 2 | 0.0536 | 0.9430 | 0.0570 |
| 3 | 0.0033 | 0.9966 | 0.0034 |
| 4 | 0.0001 | 0.9999 | 0.0001 |
sequencing redundancy 5-fold (`\rho / h = 1.7`)
| `\phi` | `P_{h,\phi} - P_{h,\phi+1}` | `1-P_{h,\phi}` | `P_{h,\phi}` |
|---|---|---|---|
| 0 | 0.4663 | 0.0000 | 1.0000 |
| 1 | 0.4114 | 0.4663 | 0.5337 |
| 2 | 0.1095 | 0.8777 | 0.1223 |
| 3 | 0.0121 | 0.9872 | 0.0128 |
| 4 | 0.0007 | 0.9993 | 0.0007 |
| 5 | 0.0000 | 1.0000 | 0.0000 |
sequencing redundancy 6-fold (`\rho / h = 2.0`)
| `\phi` | `P_{h,\phi} - P_{h,\phi+1}` | `1-P_{h,\phi}` | `P_{h,\phi}` |
|---|---|---|---|
| 0 | 0.3535 | 0.0000 | 1.0000 |
| 1 | 0.4369 | 0.3535 | 0.6465 |
| 2 | 0.1758 | 0.7904 | 0.2096 |
| 3 | 0.0309 | 0.9662 | 0.0338 |
| 4 | 0.0028 | 0.9971 | 0.0029 |
| 5 | 0.0001 | 0.9999 | 0.0001 |
sequencing redundancy 7-fold (`\rho / h = 2.3`)
| `\phi` | `P_{h,\phi} - P_{h,\phi+1}` | `1-P_{h,\phi}` | `P_{h,\phi}` |
|---|---|---|---|
| 0 | 0.2636 | 0.0000 | 1.0000 |
| 1 | 0.4264 | 0.2636 | 0.7364 |
| 2 | 0.2396 | 0.6900 | 0.3100 |
| 3 | 0.0614 | 0.9297 | 0.0703 |
| 4 | 0.0083 | 0.9911 | 0.0089 |
| 5 | 0.0006 | 0.9993 | 0.0007 |
| 6 | 0.0000 | 1.0000 | 0.0000 |
sequencing redundancy 8-fold (`\rho / h = 2.7`)
| `\phi` | `P_{h,\phi} - P_{h,\phi+1}` | `1-P_{h,\phi}` | `P_{h,\phi}` |
|---|---|---|---|
| 0 | 0.1943 | 0.0000 | 1.0000 |
| 1 | 0.3918 | 0.1943 | 0.8057 |
| 2 | 0.2902 | 0.5861 | 0.4139 |
| 3 | 0.1020 | 0.8764 | 0.1236 |
| 4 | 0.0193 | 0.9784 | 0.0216 |
| 5 | 0.0022 | 0.9977 | 0.0023 |
| 6 | 0.0002 | 0.9998 | 0.0002 |
sequencing redundancy 9-fold (`\rho / h = 3.0`)
| `\phi` | `P_{h,\phi} - P_{h,\phi+1}` | `1-P_{h,\phi}` | `P_{h,\phi}` |
|---|---|---|---|
| 0 | 0.1420 | 0.0000 | 1.0000 |
| 1 | 0.3443 | 0.1420 | 0.8580 |
| 2 | 0.3217 | 0.4864 | 0.5136 |
| 3 | 0.1480 | 0.8081 | 0.1919 |
| 4 | 0.0376 | 0.9561 | 0.0439 |
| 5 | 0.0057 | 0.9937 | 0.0063 |
| 6 | 0.0006 | 0.9994 | 0.0006 |
| 7 | 0.0000 | 1.0000 | 0.0000 |
sequencing redundancy 10-fold (`\rho / h = 3.3`)
| `\phi` | `P_{h,\phi} - P_{h,\phi+1}` | `1-P_{h,\phi}` | `P_{h,\phi}` |
|---|---|---|---|
| 0 | 0.1032 | 0.0000 | 1.0000 |
| 1 | 0.2925 | 0.1032 | 0.8968 |
| 2 | 0.3331 | 0.3958 | 0.6042 |
| 3 | 0.1933 | 0.7289 | 0.2711 |
| 4 | 0.0634 | 0.9221 | 0.0779 |
| 5 | 0.0127 | 0.9856 | 0.0144 |
| 6 | 0.0016 | 0.9982 | 0.0018 |
| 7 | 0.0001 | 0.9998 | 0.0002 |
sequencing redundancy 20-fold (`\rho / h = 6.7`)
| `\phi` | `P_{h,\phi} - P_{h,\phi+1}` | `1-P_{h,\phi}` | `P_{h,\phi}` |
|---|---|---|---|
| 0 | 0.0038 | 0.0000 | 1.0000 |
| 1 | 0.0252 | 0.0038 | 0.9962 |
| 2 | 0.0808 | 0.0290 | 0.9710 |
| 3 | 0.1633 | 0.1098 | 0.8902 |
| 4 | 0.2256 | 0.2731 | 0.7269 |
| 5 | 0.2206 | 0.4987 | 0.5013 |
| 6 | 0.1560 | 0.7194 | 0.2806 |
| 7 | 0.0811 | 0.8753 | 0.1247 |
| 8 | 0.0316 | 0.9565 | 0.0435 |
| 9 | 0.0094 | 0.9881 | 0.0119 |
| 10 | 0.0021 | 0.9974 | 0.0026 |
| 11 | 0.0004 | 0.9996 | 0.0004 |
| 12 | 0.0001 | 0.9999 | 0.0001 |
sequencing redundancy 25-fold (`\rho / h = 8.3`)
| `\phi` | `P_{h,\phi} - P_{h,\phi+1}` | `1-P_{h,\phi}` | `P_{h,\phi}` |
|---|---|---|---|
| 0 | 0.0007 | 0.0000 | 1.0000 |
| 1 | 0.0060 | 0.0007 | 0.9993 |
| 2 | 0.0247 | 0.0067 | 0.9933 |
| 3 | 0.0665 | 0.0314 | 0.9686 |
| 4 | 0.1286 | 0.0979 | 0.9021 |
| 5 | 0.1862 | 0.2266 | 0.7734 |
| 6 | 0.2052 | 0.4127 | 0.5873 |
| 7 | 0.1740 | 0.6179 | 0.3821 |
| 8 | 0.1145 | 0.7920 | 0.2080 |
| 9 | 0.0590 | 0.9065 | 0.0935 |
| 10 | 0.0241 | 0.9655 | 0.0345 |
| 11 | 0.0078 | 0.9896 | 0.0104 |
| 12 | 0.0021 | 0.9974 | 0.0026 |
| 13 | 0.0004 | 0.9995 | 0.0005 |
| 14 | 0.0001 | 0.9999 | 0.0001 |
| 15 | 0.0000 | 1.0000 | 0.0000 |
sequencing redundancy 50-fold (`\rho / h = 16.7`)
| `\phi` | `P_{h,\phi} - P_{h,\phi+1}` | `1-P_{h,\phi}` | `P_{h,\phi}` |
|---|---|---|---|
| 2 | 0.0000 | 0.0000 | 1.0000 |
| 3 | 0.0001 | 0.0000 | 1.0000 |
| 4 | 0.0006 | 0.0002 | 0.9998 |
| 5 | 0.0019 | 0.0007 | 0.9993 |
| 6 | 0.0051 | 0.0026 | 0.9974 |
| 7 | 0.0122 | 0.0077 | 0.9923 |
| 8 | 0.0250 | 0.0199 | 0.9801 |
| 9 | 0.0452 | 0.0449 | 0.9551 |
| 10 | 0.0722 | 0.0902 | 0.9098 |
| 11 | 0.1019 | 0.1623 | 0.8377 |
| 12 | 0.1273 | 0.2643 | 0.7357 |
| 13 | 0.1406 | 0.3916 | 0.6084 |
| 14 | 0.1369 | 0.5321 | 0.4679 |
| 15 | 0.1174 | 0.6690 | 0.3310 |
| 16 | 0.0887 | 0.7864 | 0.2136 |
| 17 | 0.0590 | 0.8751 | 0.1249 |
| 18 | 0.0346 | 0.9341 | 0.0659 |
| 19 | 0.0179 | 0.9687 | 0.0313 |
| 20 | 0.0082 | 0.9867 | 0.0133 |
| 21 | 0.0033 | 0.9949 | 0.0051 |
| 22 | 0.0012 | 0.9982 | 0.0018 |
| 23 | 0.0004 | 0.9995 | 0.0005 |
| 24 | 0.0001 | 0.9998 | 0.0002 |
| 25 | 0.0000 | 1.0000 | 0.0000 |
sequencing redundancy 75-fold (`\rho / h = 25.0`)
| `\phi` | `P_{h,\phi} - P_{h,\phi+1}` | `1-P_{h,\phi}` | `P_{h,\phi}` |
|---|---|---|---|
| 6 | 0.0000 | 0.0000 | 1.0000 |
| 7 | 0.0001 | 0.0000 | 1.0000 |
| 8 | 0.0002 | 0.0001 | 0.9999 |
| 9 | 0.0004 | 0.0002 | 0.9998 |
| 10 | 0.0011 | 0.0007 | 0.9993 |
| 11 | 0.0025 | 0.0018 | 0.9982 |
| 12 | 0.0052 | 0.0042 | 0.9958 |
| 13 | 0.0099 | 0.0094 | 0.9906 |
| 14 | 0.0175 | 0.0193 | 0.9807 |
| 15 | 0.0287 | 0.0367 | 0.9633 |
| 16 | 0.0436 | 0.0654 | 0.9346 |
| 17 | 0.0617 | 0.1090 | 0.8910 |
| 18 | 0.0808 | 0.1707 | 0.8293 |
| 19 | 0.0981 | 0.2515 | 0.7485 |
| 20 | 0.1101 | 0.3496 | 0.6504 |
| 21 | 0.1139 | 0.4596 | 0.5404 |
| 22 | 0.1086 | 0.5736 | 0.4264 |
| 23 | 0.0952 | 0.6821 | 0.3179 |
| 24 | 0.0767 | 0.7773 | 0.2227 |
| 25 | 0.0567 | 0.8540 | 0.1460 |
| 26 | 0.0385 | 0.9106 | 0.0894 |
| 27 | 0.0240 | 0.9491 | 0.0509 |
| 28 | 0.0137 | 0.9731 | 0.0269 |
| 29 | 0.0072 | 0.9868 | 0.0132 |
| 30 | 0.0035 | 0.9940 | 0.0060 |
| 31 | 0.0016 | 0.9974 | 0.0026 |
| 32 | 0.0006 | 0.9990 | 0.0010 |
| 33 | 0.0002 | 0.9996 | 0.0004 |
| 34 | 0.0001 | 0.9999 | 0.0001 |
| 35 | 0.0000 | 1.0000 | 0.0000 |
| 36 | 0.0000 | 1.0000 | 0.0000 |
sequencing redundancy 100-fold (`\rho / h = 33.3`)
| `\phi` | `P_{h,\phi} - P_{h,\phi+1}` | `1-P_{h,\phi}` | `P_{h,\phi}` |
|---|---|---|---|
| 11 | 0.0000 | 0.0000 | 1.0000 |
| 12 | 0.0000 | 0.0000 | 1.0000 |
| 13 | 0.0001 | 0.0001 | 0.9999 |
| 14 | 0.0002 | 0.0002 | 0.9998 |
| 15 | 0.0005 | 0.0004 | 0.9996 |
| 16 | 0.0011 | 0.0009 | 0.9991 |
| 17 | 0.0022 | 0.0020 | 0.9980 |
| 18 | 0.0040 | 0.0042 | 0.9958 |
| 19 | 0.0070 | 0.0082 | 0.9918 |
| 20 | 0.0116 | 0.0153 | 0.9847 |
| 21 | 0.0183 | 0.0269 | 0.9731 |
| 22 | 0.0273 | 0.0452 | 0.9548 |
| 23 | 0.0386 | 0.0725 | 0.9275 |
| 24 | 0.0518 | 0.1110 | 0.8890 |
| 25 | 0.0659 | 0.1628 | 0.8372 |
| 26 | 0.0793 | 0.2287 | 0.7713 |
| 27 | 0.0901 | 0.3080 | 0.6920 |
| 28 | 0.0967 | 0.3981 | 0.6019 |
| 29 | 0.0976 | 0.4948 | 0.5052 |
| 30 | 0.0927 | 0.5924 | 0.4076 |
| 31 | 0.0827 | 0.6851 | 0.3149 |
| 32 | 0.0693 | 0.7679 | 0.2321 |
| 33 | 0.0544 | 0.8371 | 0.1629 |
| 34 | 0.0400 | 0.8915 | 0.1085 |
| 35 | 0.0276 | 0.9315 | 0.0685 |
| 36 | 0.0178 | 0.9591 | 0.0409 |
| 37 | 0.0108 | 0.9769 | 0.0231 |
| 38 | 0.0061 | 0.9877 | 0.0123 |
| 39 | 0.0033 | 0.9938 | 0.0062 |
| 40 | 0.0016 | 0.9971 | 0.0029 |
| 41 | 0.0008 | 0.9987 | 0.0013 |
| 42 | 0.0003 | 0.9994 | 0.0006 |
| 43 | 0.0001 | 0.9998 | 0.0002 |
| 44 | 0.0001 | 0.9999 | 0.0001 |
| 45 | 0.0000 | 1.0000 | 0.0000 |
| 46 | 0.0000 | 1.0000 | 0.0000 |
Beyond Belief Campaign BRCA Art
Fuelled by philanthropy, findings into the workings of BRCA1 and BRCA2 genes have led to groundbreaking research and lifesaving innovations to care for families facing cancer.
This set of 100 one-of-a-kind prints explore the structure of these genes. Each artwork is unique — if you put them all together, you get the full sequence of the BRCA1 and BRCA2 proteins.

Propensity score weighting
The needs of the many outweigh the needs of the few. —Mr. Spock (Star Trek II)
This month, we explore a related and powerful technique to address bias: propensity score weighting (PSW), which applies weights to each subject instead of matching (or discarding) them.
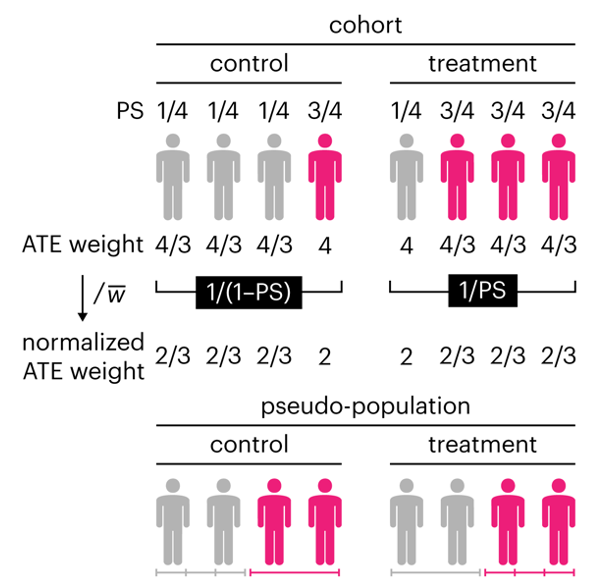
Kurz, C.F., Krzywinski, M. & Altman, N. (2025) Points of significance: Propensity score weighting. Nat. Methods 22:1–3.
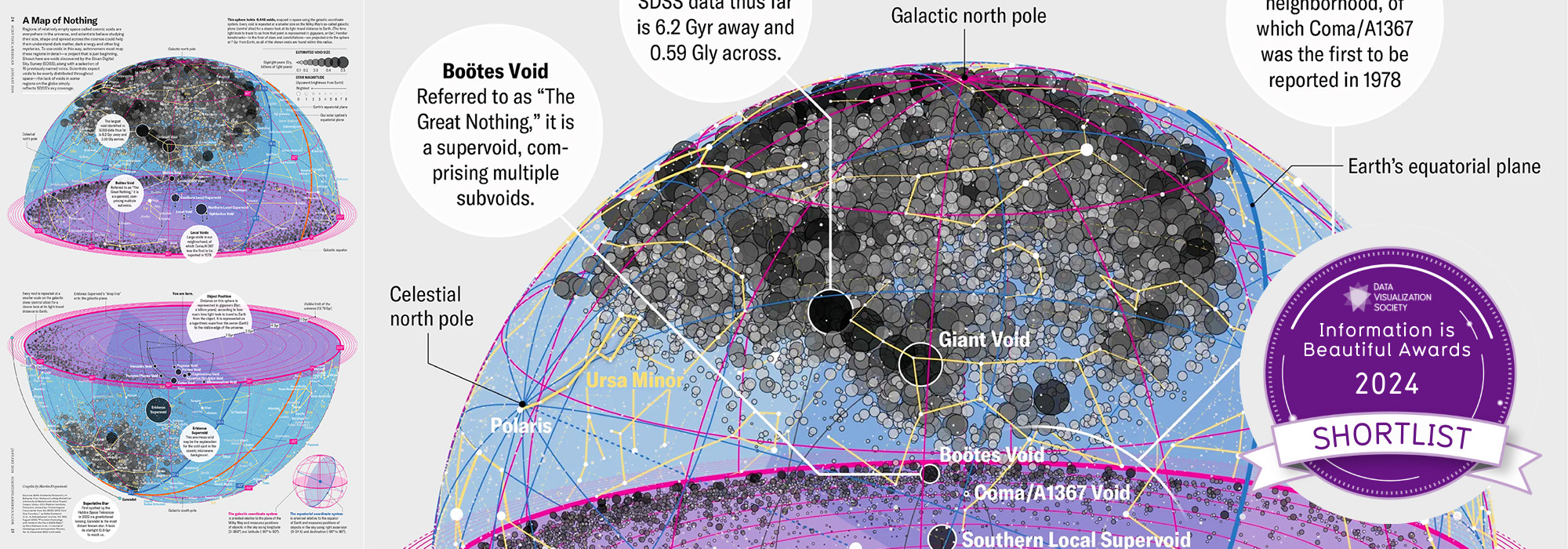
Happy 2025 π Day—
TTCAGT: a sequence of digits
Celebrate π Day (March 14th) and sequence digits like its 1999. Let's call some peaks.
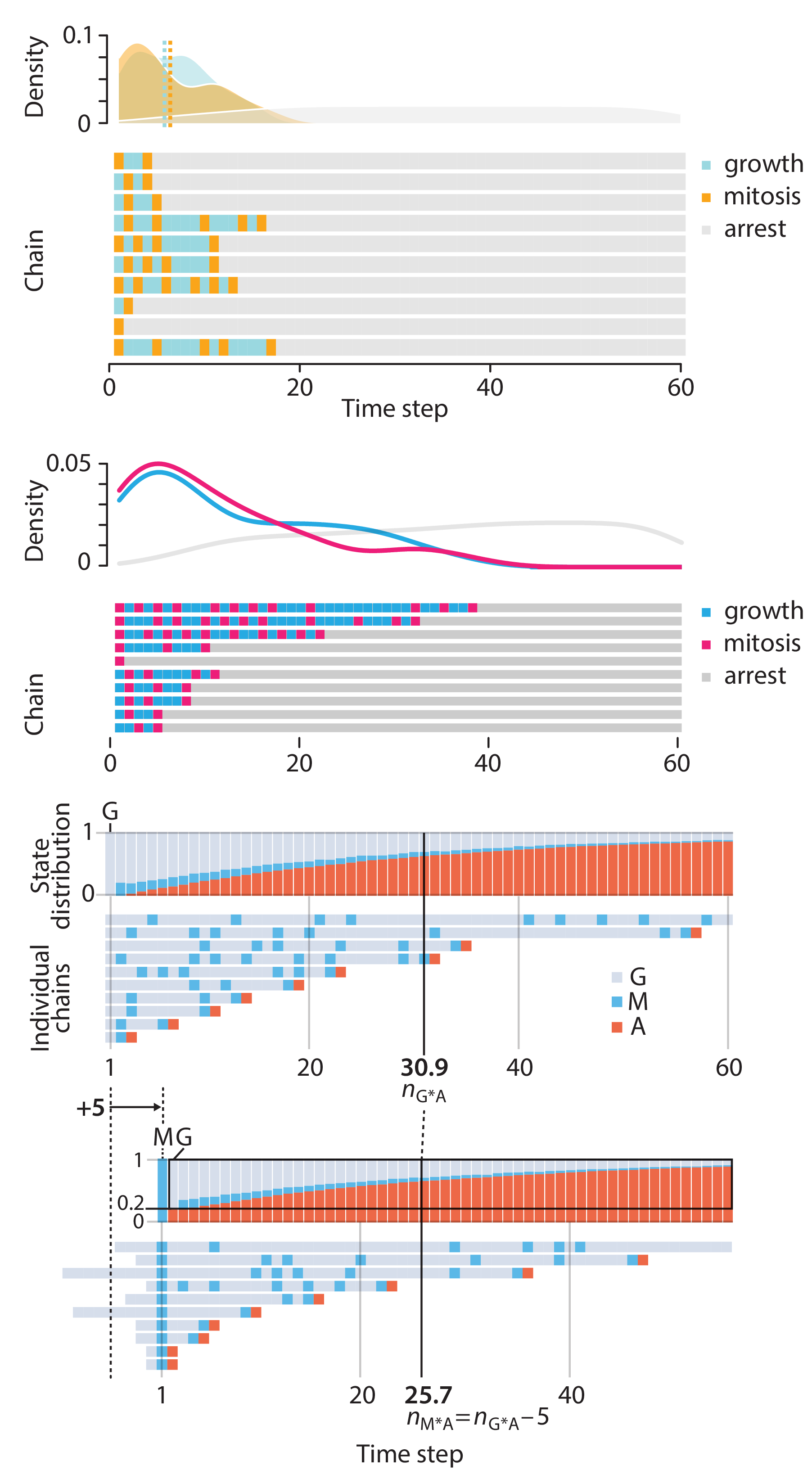
Crafting 10 Years of Statistics Explanations: Points of Significance
I don’t have good luck in the match points. —Rafael Nadal, Spanish tennis player
Points of Significance is an ongoing series of short articles about statistics in Nature Methods that started in 2013. Its aim is to provide clear explanations of essential concepts in statistics for a nonspecialist audience. The articles favor heuristic explanations and make extensive use of simulated examples and graphical explanations, while maintaining mathematical rigor.
Topics range from basic, but often misunderstood, such as uncertainty and P-values, to relatively advanced, but often neglected, such as the error-in-variables problem and the curse of dimensionality. More recent articles have focused on timely topics such as modeling of epidemics, machine learning, and neural networks.
In this article, we discuss the evolution of topics and details behind some of the story arcs, our approach to crafting statistical explanations and narratives, and our use of figures and numerical simulations as props for building understanding.
Altman, N. & Krzywinski, M. (2025) Crafting 10 Years of Statistics Explanations: Points of Significance. Annual Review of Statistics and Its Application 12:69–87.
Propensity score matching
I don’t have good luck in the match points. —Rafael Nadal, Spanish tennis player
In many experimental designs, we need to keep in mind the possibility of confounding variables, which may give rise to bias in the estimate of the treatment effect.
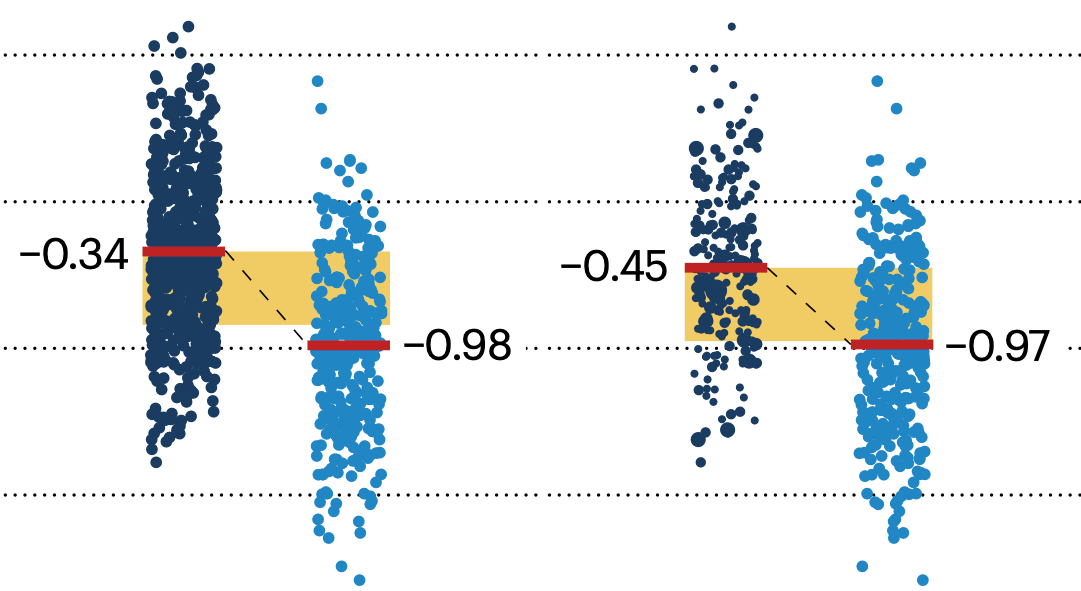
If the control and experimental groups aren't matched (or, roughly, similar enough), this bias can arise.
Sometimes this can be dealt with by randomizing, which on average can balance this effect out. When randomization is not possible, propensity score matching is an excellent strategy to match control and experimental groups.
Kurz, C.F., Krzywinski, M. & Altman, N. (2024) Points of significance: Propensity score matching. Nat. Methods 21:1770–1772.
Understanding p-values and significance
P-values combined with estimates of effect size are used to assess the importance of experimental results. However, their interpretation can be invalidated by selection bias when testing multiple hypotheses, fitting multiple models or even informally selecting results that seem interesting after observing the data.
We offer an introduction to principled uses of p-values (targeted at the non-specialist) and identify questionable practices to be avoided.
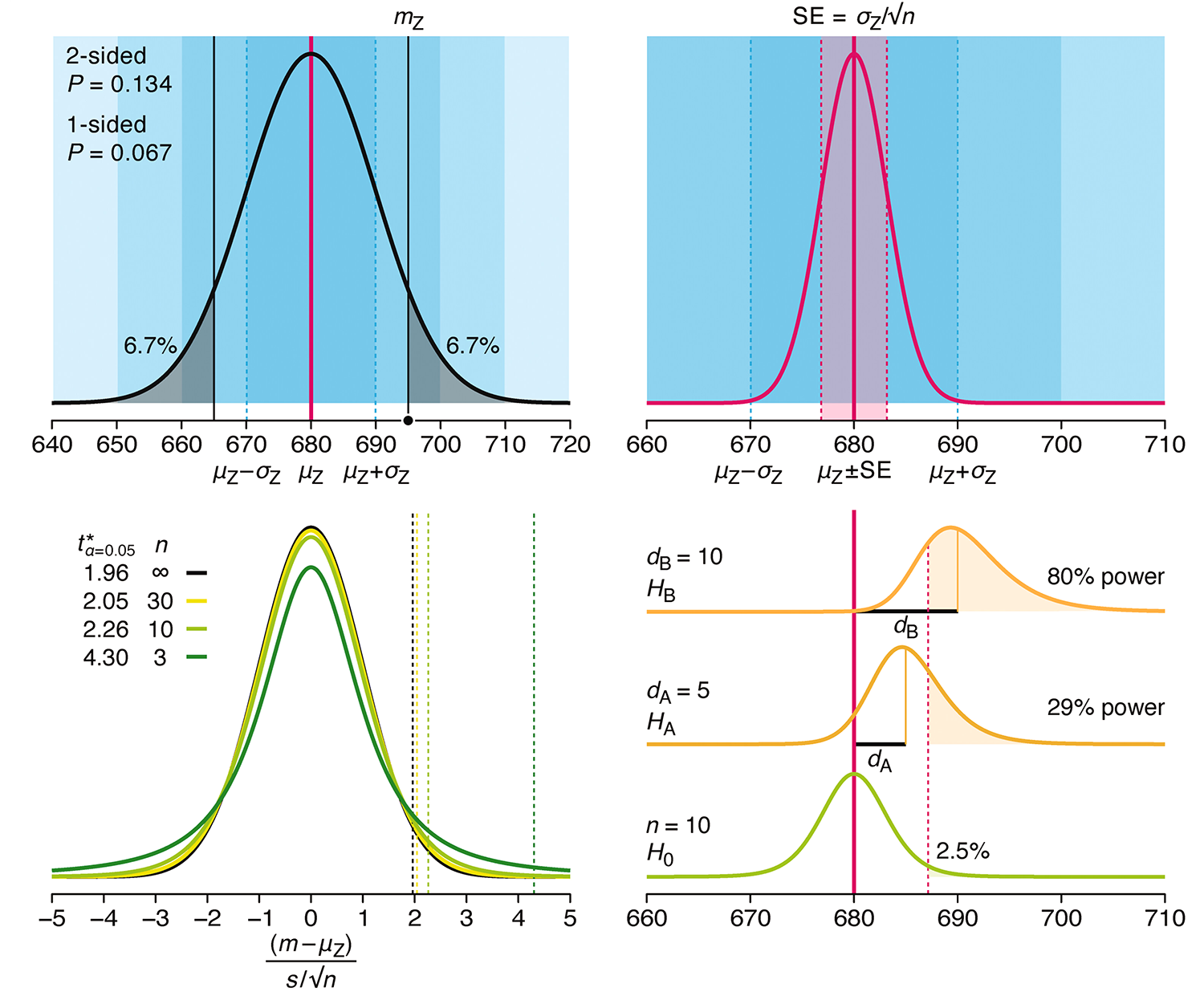
Altman, N. & Krzywinski, M. (2024) Understanding p-values and significance. Laboratory Animals 58:443–446.