
science + genomics
What if we were to print what we sequence?
Expressing the amount of sequence in the human genome in terms of the number of printed pages has been done before. At the Broad Institute, all of the human reference genome is printed in bound volumes.
At our sequencing facility, we sequence about 1 terabases per day. This is equivalent to 167 diploid human genomes (167 × 6 gigabases). The sequencing is done using a pool of 13 Illumina HiSeq 2500 sequencers, of which about 50% are sequencing at any given time.

This sequencing is extremely fast.
To understand just how fast this is, consider printing this amount of sequence using a modern office laser printer. Let's pick the HP P3015n which costs about $400—a cheap and fast network printer. It can print at about 40 pages per minute.
If we print the sequence at 6pt Courier using 0.25" margins, each 8.5" × 11" page will accomodate 18,126 bases. I chose this font size because it's reasonably legible. To print 1 terabases we need `10^12 / 18126 = 55.2` million pages.
If we print continuously at 40 pages per minute, we need `10^12 / (18126*40*1440) = 957.8` days.
If we had 958 printers working around the clock, we could print everything we sequence and not fall behind. This does not account for time required to replenish toner or paper.
what's cheaper, sequencing or printing?
It costs us about $12,000 to sequence a terabase in reagents. If we do it on a cost-recovery basis, it is about twice that, to include labor and storage. Let's say $25,000 per terabase.
Coincidentally, this is about $150 per 1× coverage of a diploid human genome. The cost of sequencing a single genome would be significantly higher because of overhead. To overcome gaps in coverage and to be sensitive to alleles in heterogenous samples, sequencing should be done to 30× or more. For example, we sequence cancer genomes at over 100×. For theory and review see Aspects of coverage in medical DNA sequencing by Wendl et al. and Sequencing depth and coverage: key considerations in genomic analyses by Sims et al.. (Thanks to Nicolas Robine for pointing out that redundant coverage should be mentioned here).
Printing is 44× more expensive than sequencing, per base: 25 n$ vs 1.1 μ$.
I should mention that the cost of analyzing the sequenced genome should be considered—this step is always the much more expensive one. In The $1,000 genome, the $100,000 analysis? Mardis asks "If our efforts to improve the human reference sequence quality, variation, and annotation are successful, how do we avoid the pitfall of having cheap human genome resequencing but complex and expensive manual analysis to make clinical sense out of the data?"
The cost of a single printed page (toner, power, etc) is about $0.02–0.05, depending on the printer. Let's be generous and say it's $0.02. To print 55.2 million pages would cost us $1.1M. This is about 44 times as expensive as sequencing.

Think about this. It's 44× more expensive to merely print a letter on a page than it is to determine it from the DNA of a cell. In other words, to go from the physical molecule to a bit state on a disk is much cheaper than from a bit state on a disk to a representation of the letter on a page.
Per base, our sequencing costs `$25000/10^12 = $25*10^-9`, or 25 nanodollars. At $0.02 and 18,126 bp per page, printing costs `0.02/18126 = $1.1*10^-6` or 1.1 microdollars.
If at this point you're thinking that printing isn't practical, the fact that the pages would weigh 248,000 kg and stack to 5.5 km should cinch the argument.
The capital cost of sequencing is, of course, much higher. The printers themselves would cost about $400,000 to purchase. The 6 sequencers, on the other hand, cost about $3,600,000.
sequencing is as fast as downloading
We sequence at a rate close to the average internet bandwidth available to the public.
At 3.86 Mb/s, we could download a terabase of compressed sequence in a day, assuming the sequence can be compressed by a factor of 3. This level of compression is reasonable—the current human assembly is 938 Mb zipped).
In other words, you would have to be downloading essentially continuously to keep up with our sequencing.
Propensity score weighting
It is not certain that everything is uncertain. —Blaise Pascal
We have already explored how we can mitigate bias caused by confounding variables in observational studies using propensity score (PS) matching (PSM) and propensity score weighting (PSW). However, any statistical model is only as good as its assumptions and, if it is specified incorrectly, it can itself produce biased estimates of the treatment effect.
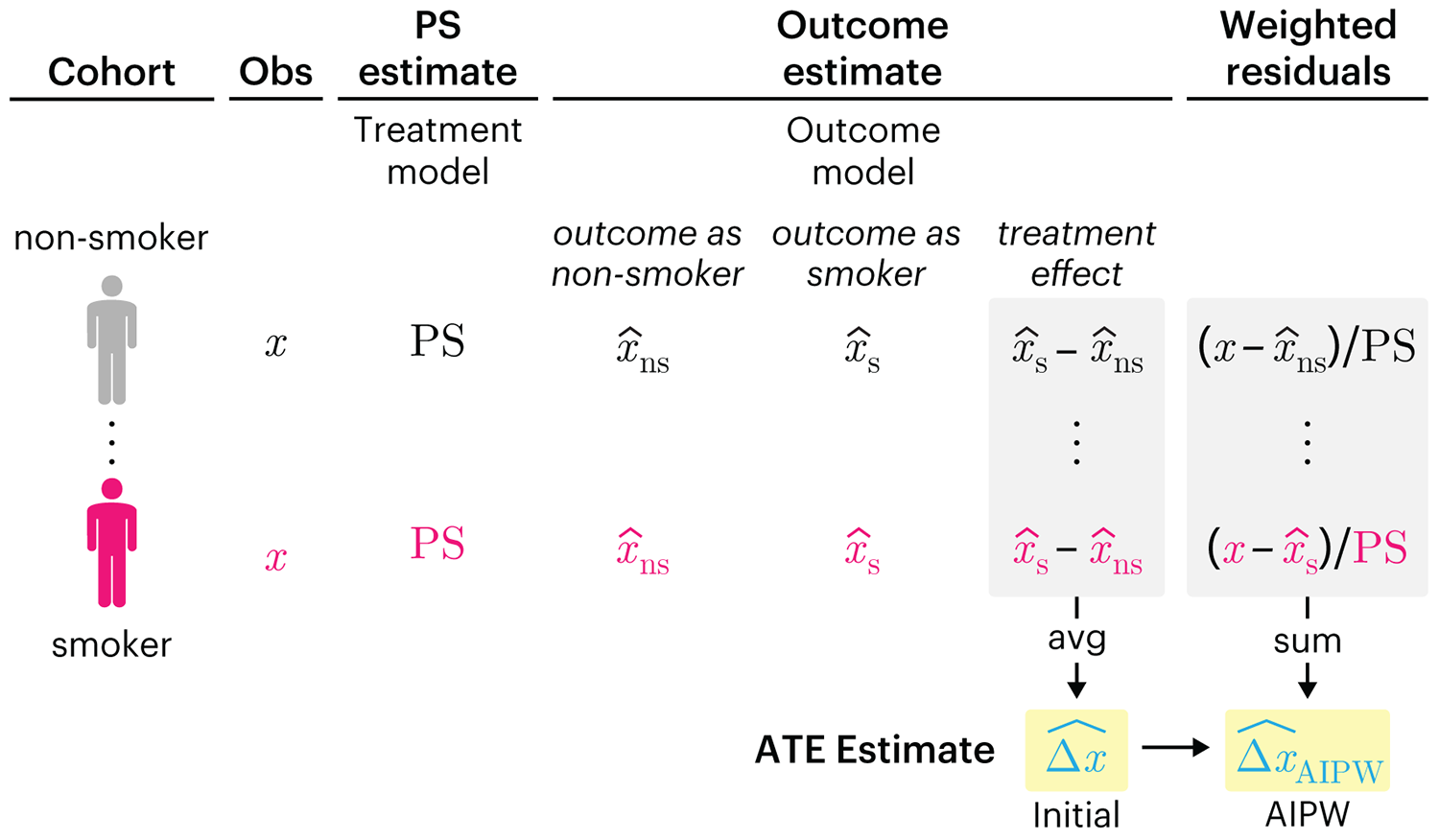
This month, we explore double robustness, a powerful statistical concept that provides a valuable “safety net” against the risk of an incorrect model. It offers two opportunities, instead of just one, to obtain a valid estimate of the treatment effect — making it possible to draw credible causal inferences from observational data without having to depend on a single set of modeling assumptions.
Kurz, C.F., Krzywinski, M. & Altman, N. (2026) Points of significance: Double Robustness. Nat. Methods 23:868–869.
Nature Biotechnology cover
My cover design on the 7 April 2026 Nature Biotechnology issue shows the dendrogram that represents a cluster of uniquely expressed (or downregulated) genes in human naive stem cells induced from such cells. Within each dendrogram block, the genomic barcode sequence (sampled from Supplementary Table 1) is depicted with a Code 39 barcode. The highlighted barcode is one of those used for cell isolation.
Ishiguro S. et al. A multi-kingdom genetic barcoding system for precise clone isolation (2026) Nature Biotechnology 44:616–629.

Browse my gallery of cover designs.

Happy 2026 π Day—
Art for the 5%
Celebrate π Day (March 14th) and enjoy the art — but only if you're part of the 5%.
Go ahead, see what you can't see.

Ishihara's Tests for Colour Deficiency
Authentic and accurate images of Ishihara's test plates photographed (and lovingly color-corrected) from the 38-plate Ishihara's Tests for Colour Deficiency.
I also provide the position, size, and color of each circle on each test plate.


Symmetric alternatives to the ordinary least squares regression
What immortal hand or eye, could frame thy fearful symmetry? — William Blake, "The Tyger"
This month, we look at symmetric regression, which, unlike simple linear regression, it is reversible — remaining unaltered when the variables are swapped.
Simple linear regression can summarize the linear relationship between two variables `X` and `Y` — for example, when `Y` is considered the response (dependent) and `X` the predictor (independent) variable.
However, there are times when we are not interested (or able) to distinguish between dependent and independent variables — either because they have the same importance or the same role. This is where symmetric regression can help.

Luca Greco, George Luta, Martin Krzywinski & Naomi Altman (2025) Points of significance: Symmetric alternatives to the ordinary least squares regression. Nat. Methods 22:1610–1612.