Learning Circos
Bioinformatics and Genome Analysis
Institut Pasteur Tunis Tunisia, December 10 – December 11, 2018
v2.00 6 Dec 2018 Download PDF slides
v2.00 6 Dec 2018
A 2- or 4-day practical mini-course in Circos, command-line parsing and scripting. This material is part of the Bioinformatics and Genome Analysis course held at the Institut Pasteur Tunis.
Quick links
BCGA 2018 | 1-day Circos course | Circos documentation best practices getting started | Brewer palette swatches | Color resources | Nature Methods Points of View Points of Significance
Getting started with Circos: Yeast and Ebola
Monday 10 December 2018 — Day 1
9h00 - 10h30 | Lecture 1 — Introduction to Circos
11h00 - 12h30 | Lecture (practical) 2 — Visualizing gene distribution and size in Yeast: the histogram data track
14h00 - 15h30 | Lecture (practical) 3 — Conservation in Yeast: the link data track
16h00 - 18h00 | Lecture (practical) 4 — Visualizing an Ebola strain
Concepts covered today
Circos configuration, common Circos errors, Circos debugging, ideograms, selecting ideograms with regular expressions, data tracks, histograms, links, downloading files from UCSC genome browser, essential command-line tools and basic scripting, using bash to create data files for Ebola genome strains, color definitions, using transparency, Brewer palettes, runtime formatting rules, accessing data track statistics, input data formats
Introduction to Circos
This lecture covers the file organization of my lectures, how to use the files and some commonly seen error messages generated by Circos that you might encounter on the way.
You can follow this material on any computer that has Circos installed. You can always install it yourself, too.
http://www.circos.ca/documentation/tutorials/configuration/
What you will learn
1. how Circos works
2. how to visualize various data with Circos
3. reinforce your command line skills
Finally, you will create a unique image montage as a souvenir image to finish off the course!
How Circos works
Circos does not have an interface.
Circos is a command line program.
Circos takes plain-text files as input, which define all aspects of the image.
Circos configuration
A very basic configuration file looks like this
karyotype = ../../data/karyotype.txt
Everything below is details that we'll worry about later.
chromosomes_units = 100
<<include ideogram.conf>>
<<include ../../../etc/ticks.conf>>
<<include ../../../etc/image.conf>>
<<include etc/colors_fonts_patterns.conf>>
<<include etc/housekeeping.conf>>
The karyotype.txt file defines the name and length of all the segments placed around the circle.
For example, you'll start off by drawing some data on three different Yeast strains, for which the karyotype file is
# chr - name label 0 length color
chr - sace-a saceA 0 230218 grey
chr - sace-b saceB 0 813184 grey
chr - sace-d saceD 0 1531933 grey
...
chr - cagl-a caglA 0 491328 orange
chr - cagl-b caglB 0 502101 orange
chr - cagl-c caglC 0 558804 orange
...
chr - zyro-a zyroA 0 1114666 blue
chr - zyro-b zyroB 0 1388208 blue
chr - zyro-c zyroC 0 1464093 blue
...
You don't have to draw all the segments defined in the file. For example, by using the chromosomes parameter you can choose to draw only one segment.
chromosomes = sace-a
To draw data segments, you first create a plain-text file
cagl-a 0 19999 1
cagl-a 20000 39999 10
cagl-a 40000 59999 5
...
and then ask Circos to draw the data using <plot> blocks.
<plots>
<plot>
file = ../../data/genes.count.20kb.txt
type = histogram
</plot>
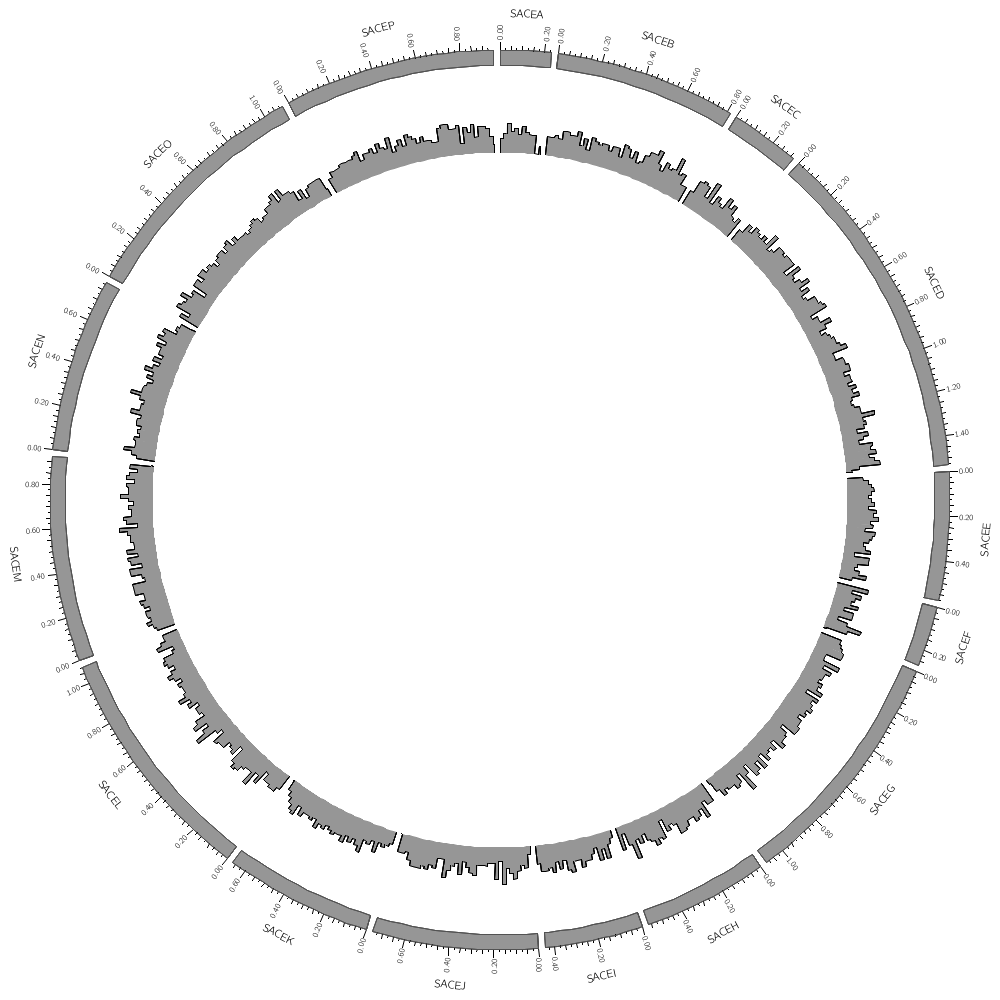
You can add as many plot tracks as you like — they can be placed anywhere in the image. They can even overlap.
To do this, just add another plot block.
<plots>
<plot>
file = ../../data/genes.count.20kb.txt
type = histogram
r1 = 0.95r
r0 = 0.80r
<backgrounds>
<background>
color = vlgrey
</background>
</backgrounds>
</plot>
<plot>
file = ../../data/genes.avgsize.10kb.txt
type = histogram
r1 = 0.79r
r0 = 0.65r
fill_color = blue
</plot>
</plots>
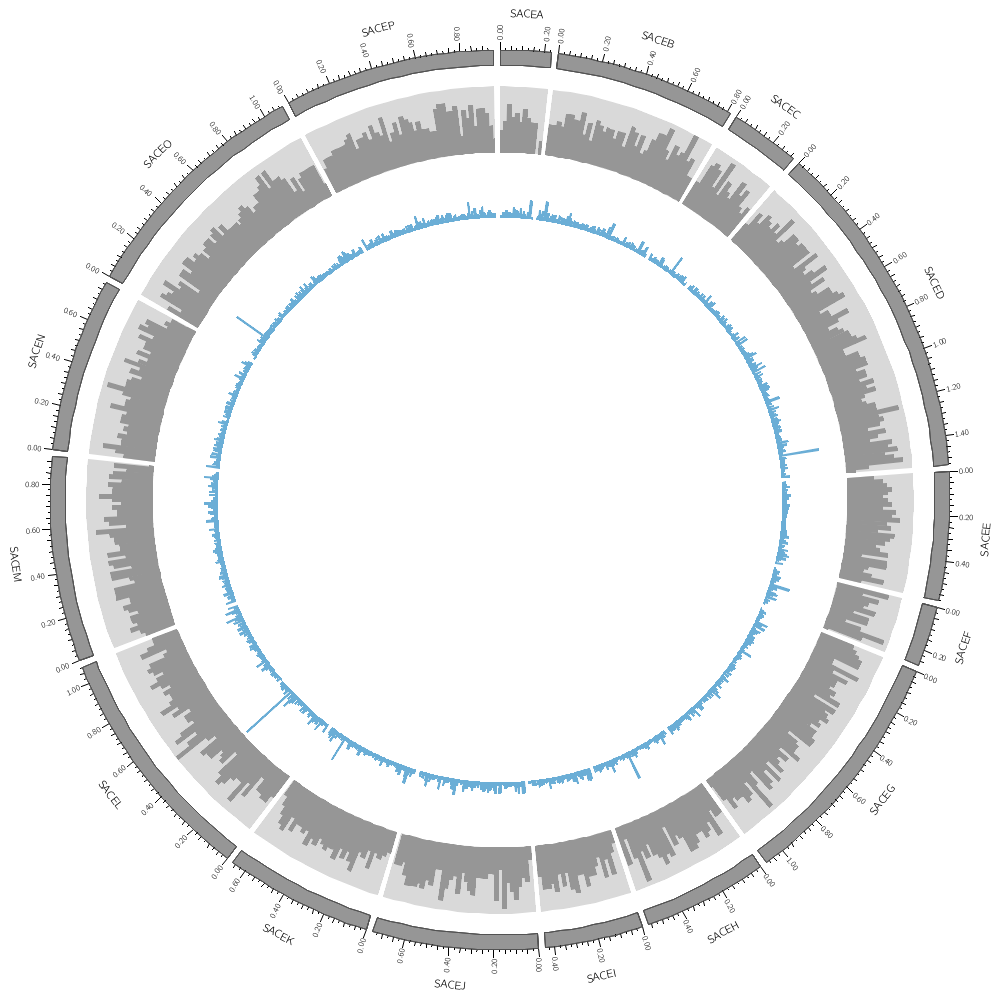
The data files define tracks for any number of segments. In our example, we have data on each segment.
This is the image that you'll be creating in your first practical session. There are many details that go into creating the image such as
1. position and order of segments
2. segment thickness and labels
3. tick and tick labels
We'll skip past these details for now so that you can get started quickly.
parsing data on the command line
You're also going to be spending a lot of time on the command line parsing data, asking statistical questions and preparing Circos input files.
cat - listing a file
grep - matching lines with regular expressions
sed - replacing strings
cut - pulling out fields
sort - sorting lines
uniq - counting unique lines
awk - robust data extraction and reporting tool, particularly useful for rearranging fields
Command line parsing patterns
List lines from file.txt with top 10 largest values in column 3
> awk '{print $3,$0}' file.txt | sort -nr | head -10 | cut -d " " -f 2-
List all lines that match neither "abc" nor "def" and replace all instances of "chr" with "hs"
> grep -v 'abc\|def' file.txt | sed 's/chr/hs/g'
List all lines except comment or blank lines
> egrep -v '^($|#)' file.txt
Circos tools
Circos also comes with helper scripts that achieve common tasks.
Because Circos does not perform analysis (or calculations) you will need to generate data sets sampled at the right resolution.
For example, the resample tool aggregates data in bins and reports statistics like average, min, max and count.
So you can take a list of gene positions and their sizes and create a count of genes in each 10kb window.
sace-a 1807 2169 363 name=YAL068C
sace-a 2480 2707 228 name=YAL067W-A
sace-a 7235 9016 1782 name=YAL067C
sace-a 11565 11951 387 name=YAL065C
sace-a 12046 12426 381 name=YAL064W-B
sace-a 13363 13743 381 name=YAL064C-A
sace-a 21566 21850 285 name=YAL064W
sace-a 22395 22685 291 name=YAL063C-A
...
cat genes.txt | $DIR/resample/bin/resample -bin 10000 -count
sace-a 0 9999 3
sace-a 10000 19999 3
sace-a 20000 29999 3
...
You can script this to generate counts (and other statistics) for various windows.
for binkb in 50 75 100 ; do
binsize=$((1000*binkb))
cat ../../data/genes.txt | $CTOOLS/resample/bin/resample -bin $binsize -count > genes.count.${binkb}kb.txt
cat ../../data/genes.txt | $CTOOLS/resample/bin/resample -bin $binsize -avg > genes.avgsize.${binkb}kb.txt
done
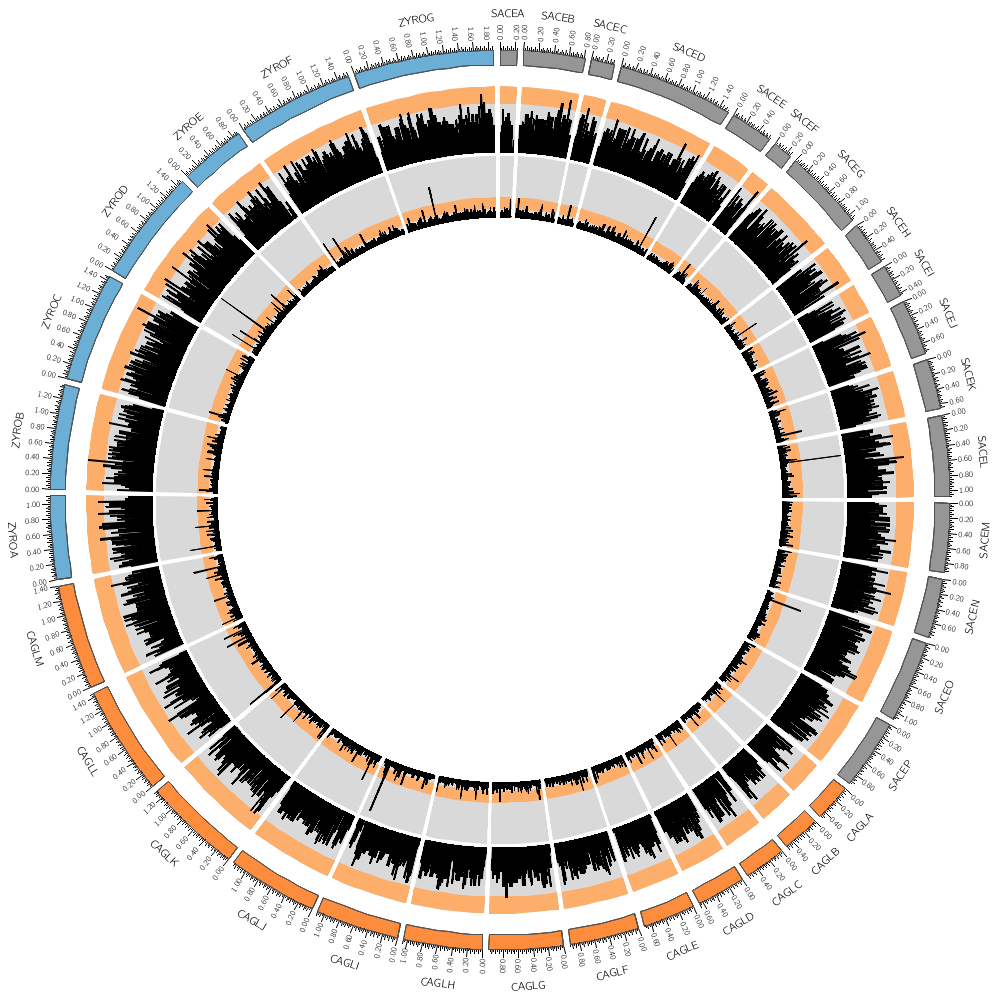
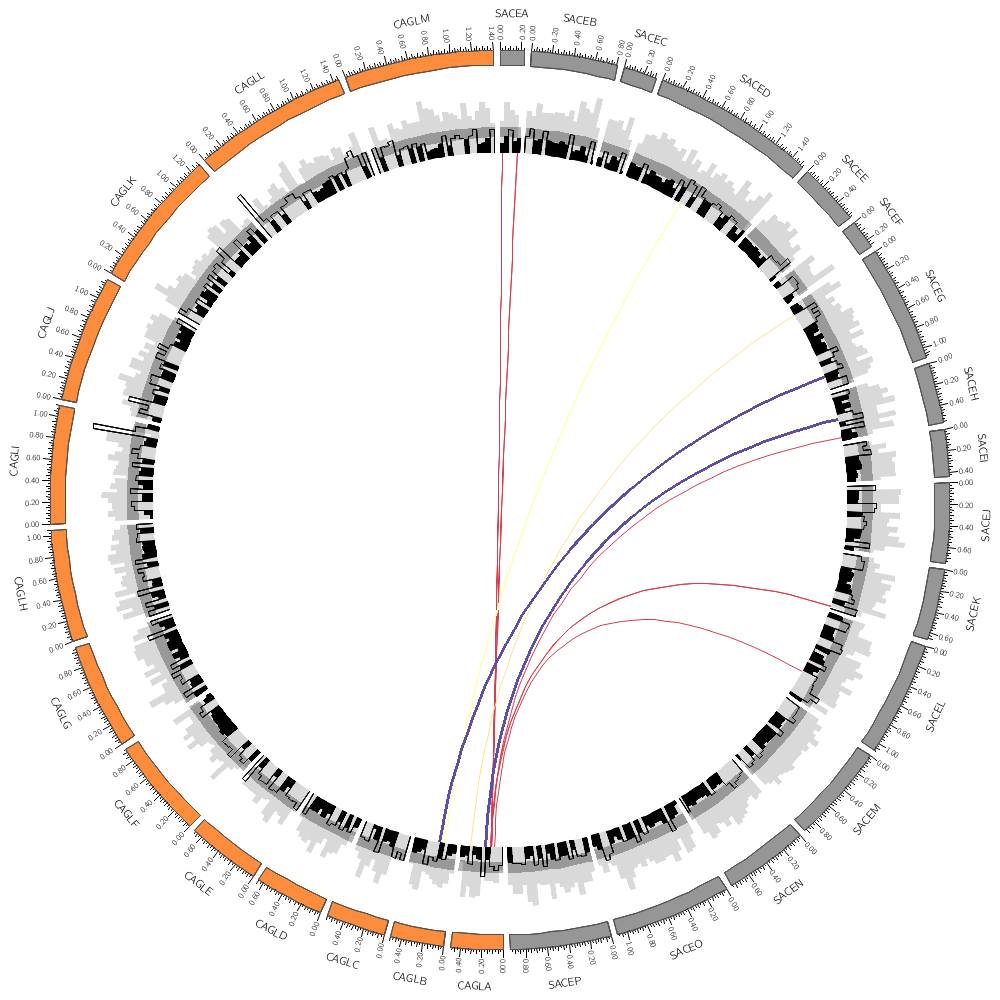
Drawing links
In today's Lecture 3 you will learn how to draw links.
These can represent any relationship between genome positions, such as conservation.
The format of a link file is simple: a pair of coordinates.
cagl-a 19487 21757 sace-g 450197 452104
cagl-a 22260 24440 sace-g 452404 454560
cagl-a 25241 27622 sace-b 207194 209224
...
And links are defined in circos.conf using a <link> block.
<links>
<link>
file = ../../data/link_cagl_sace.txt
radius = 0.98r
...
</link>
</links>
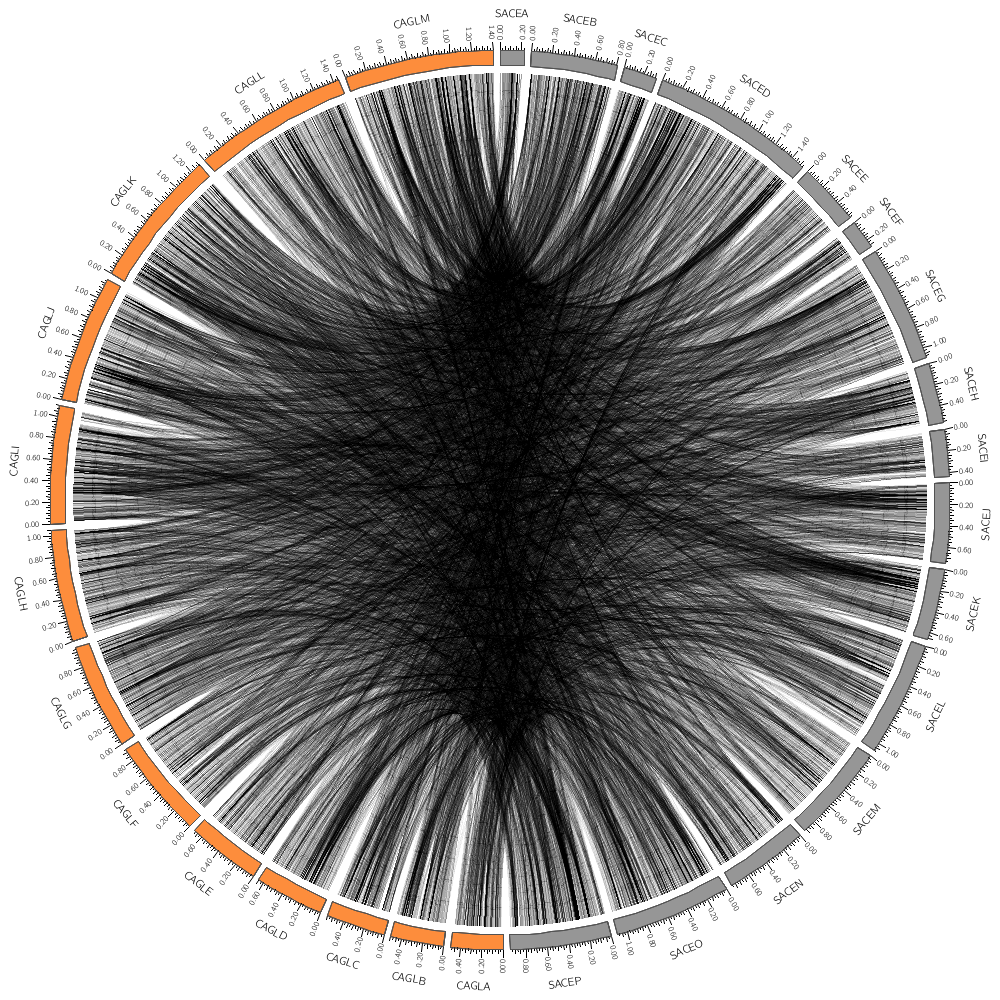
For example, you'll learn how to limit which links are drawn (e.g. by the size of the conserved area) and color them based on the size.
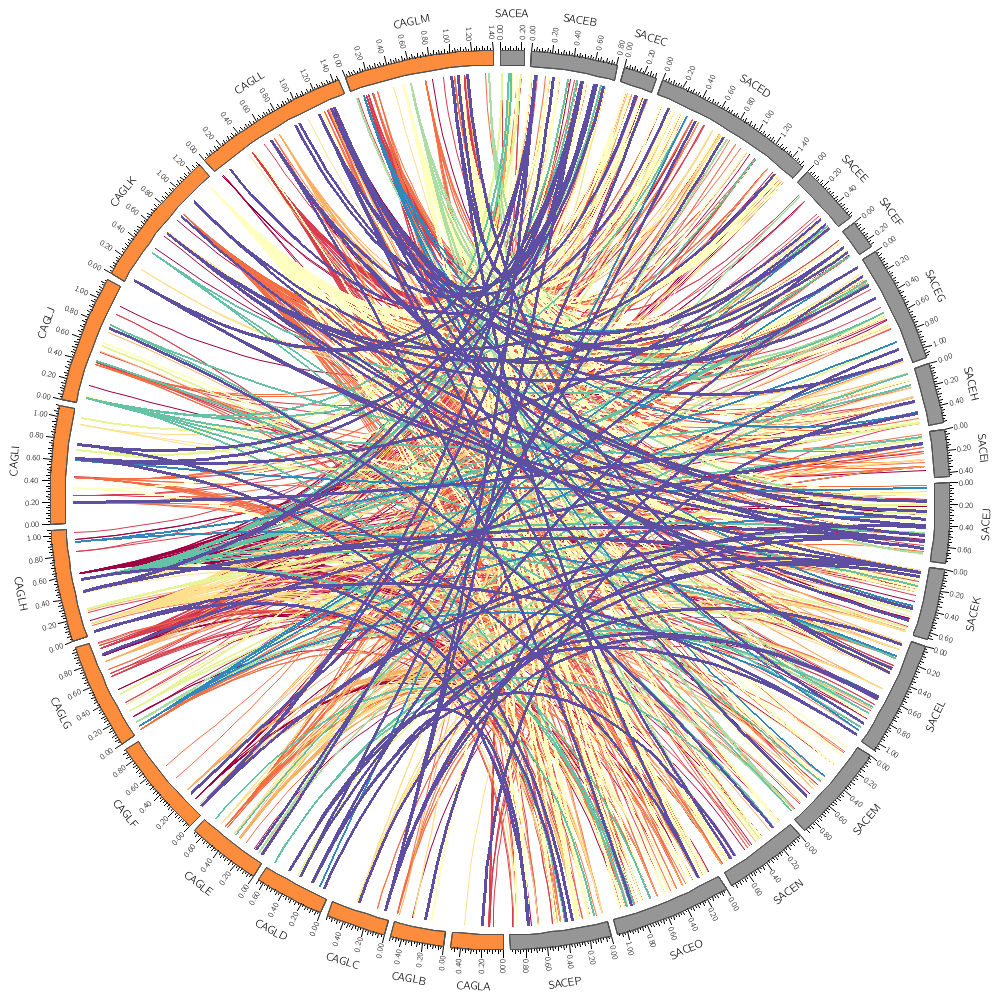
Rules
Circos allows you to write simple rules to determine whether and how data should be drawn. These can apply to <plot> and <link> blocks.
<link>
...
<rules>
<rule>
condition = var(size1) < 4000
show = no
</rule>
<rule>
#... another rule
</rule>
<rule>
#... another rule
</rule>
</rules>
</link>
Each rule has a condition. Every data point is tested and if the condition is true, the rule applies to the data point and no more rules are tested for that data point (unless you specifically specify that they shoudl). If the condition is not true, subsequent rules are tested.
The rules reference the values and formatting of a data point using var(X) where X is a property like chr, start, end, color and so on.
For links, because they are a pair of coordinates, we have chr1, start1 and end1 for the first coordinate and chr2, start2 and end2 for the second.
Color encodings
Color is a very useful way to encode information about data.
Circos supports very flexible color definitions.
There are many helpful color names already defined, like
black
grey
red
orange
yellow
blue
green
purple
Each color has a light and dark variant
vvdred very very dark red
vdred very dark red
dred dark red
red red
lred light red
vlred very light red
vvlred very very light red
Pretty funny, eh?
Brewer palettes
Brewer palettes are predefined perceptually uniform color schemes for encoding quantitative and qualitative data.

You can find all the swatches for the palette in handouts/brewer-palettes-swatches.pdf or on my Brewer palette resource page.
For example, let's look at two diverging palettes: spectral and pink-yellow-green.
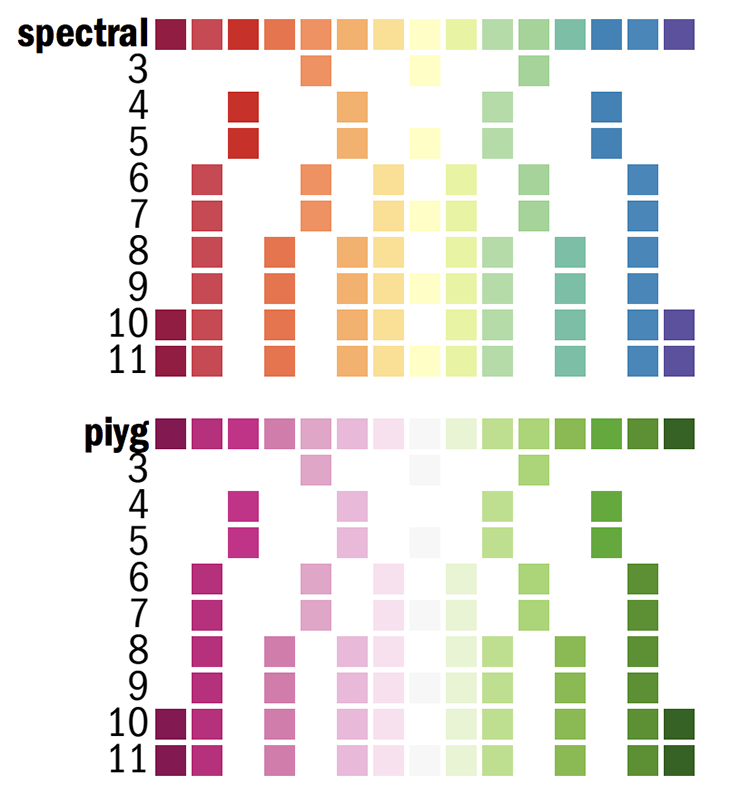
Each of the k=1..N colors for the N-color palette is defined as
spectral-N-div-k
piyg-N-div-k
For example spectral-5-div-1 is the first color (dark red) in the 5-color spectral palette.
The individual color definitions for red mentioned above are actually based on the Brewer palettes.

vvlred = reds-7-seq-1
vlred = reds-7-seq-2
lred = reds-7-seq-3
red = reds-7-seq-4
dred = reds-7-seq-5
vdred = reds-7-seq-6
vvdred = reds-7-seq-7
Creating input data files
A common question is "I have a fasta file, how do I visualize it?"
Let's take a moment to think about why this question cannot have just one answer.
In the last lecture today, you'll be creating data files based on one of the Ebola assemblies.
ebola.assembly.txt
#bin chrom chromStart chromEnd ix type frag fragStart fragEnd strand
585 KM034562v1 0 18957 1 F KM034562.1 0 18957 +
You'll use the assembly file to create a karyotype file
ebola.karyotype.txt
chr - ebola ebola 0 18957 black
You'll also parse the gene file
ebola.genes.txt
#bin name chrom strand txStart txEnd cdsStart cdsEnd exonCount exonStarts exonEnds
585 NP KM034562v1 + 55 3026 469 2689 1 55, 3026,
585 VP35 KM034562v1 + 3031 4407 3128 4151 1 3031, 4407,
585 VP40 KM034562v1 + 4389 5894 4478 5459 1 4389, 5894,
585 GP KM034562v1 + 5899 8305 6038 8068 2 5899,6922, 6920,8305,
585 sGP KM034562v1 + 5899 8305 6038 7133 1 5899, 8305,
585 ssGP KM034562v1 + 5899 8305 6038 6933 2 5899,6923, 6922,8305,
585 VP30 KM034562v1 + 8287 9740 8508 9375 1 8287, 9740,
585 VP24 KM034562v1 + 9884 11518 10344 11100 1 9884, 11518,
585 L KM034562v1 + 11500 18282 11580 18219 1 11500, 18282,
To create a track file that can be used to draw the position of the genes.
ebola.genes.circos.txt
ebola 55 3026 1 name=NP
ebola 3031 4407 1 name=VP35
ebola 4389 5894 1 name=VP40
ebola 5899 8305 2 name=GP
ebola 5899 8305 1 name=sGP
ebola 5899 8305 2 name=ssGP
ebola 8287 9740 1 name=VP30
ebola 9884 11518 1 name=VP24
ebola 11500 18282 1 name=L
Finally, you'll take the variation file
ebola.variation.txt
#chrom chromStart chromEnd name score strand thickStart thickEnd reserved gene type hgsv blosum62 countInNew freqInNew
KM034562v1 126 127 C/T 0 + 126 127 0,0,0 NP noncoding NA 81 1.000000
KM034562v1 154 155 A/C 0 + 154 155 0,0,0 NP noncoding NA 81 1.000000
KM034562v1 181 182 A/G 0 + 181 182 0,0,0 NP noncoding NA 81 1.000000
KM034562v1 186 187 A/G 0 + 186 187 0,0,0 NP noncoding NA 81 1.000000
KM034562v1 235 236 T/C 0 + 235 236 0,0,0 NP noncoding NA 81 1.000000
KM034562v1 256 257 A/G 0 + 256 257 0,0,0 NP noncoding NA 81 1.000000
KM034562v1 260 261 C/T 0 + 260 261 0,0,0 NP noncoding NA 81 1.000000
and create a file like this
ebola 126 127 1.000000 snp=C/T,gene=NP
ebola 154 155 1.000000 snp=A/C,gene=NP
ebola 181 182 1.000000 snp=A/G,gene=NP
ebola 186 187 1.000000 snp=A/G,gene=NP
ebola 235 236 1.000000 snp=T/C,gene=NP
ebola 256 257 1.000000 snp=A/G,gene=NP
...
ebola 2913 2914 1.000000 snp=T/G,gene=noncoding
ebola 2932 2933 1.000000 snp=T/C,gene=noncoding
ebola 3083 3084 1.000000 snp=C/A,gene=VP35
ebola 3115 3116 0.024691 snp=C/G,gene=VP35
...
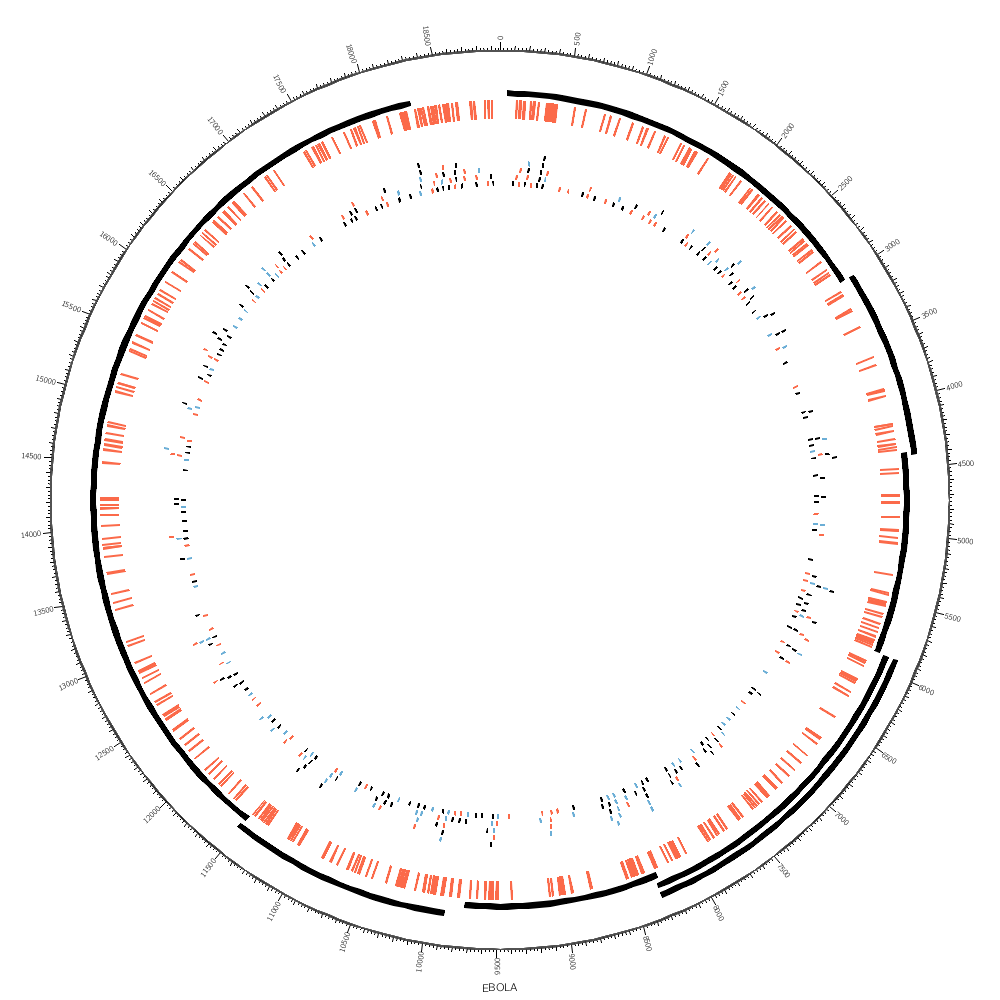
You'll see how you can use highlight and tile tracks to show regions and elements.
You'll see how rules can be used to color the SNPs, which are in the tile track. Since each SNP has a parameter snp (see the file above), you can test the value with a regular expression.
<rule>
condition = var(snp) =~ /^A/
color = red
</rule>
<rule>
condition = var(snp) =~ /A$/
color = blue
</rule>
Day 2
Tomorrow, you're going to be working on a series of more complicated data parsing challenges.
You have already seen how to draw data: histograms, tiles, links and so on. It's even more important that you are comfortable with parsing and formatting data files.
Human genome
Your first challenge will be to explore the karyotypes that come with Circos and draw the chromosomes of the human genome.
karyotype.human.txt
chr - hs1 1 0 249250621 chr1
chr - hs2 2 0 243199373 chr2
chr - hs3 3 0 198022430 chr3
chr - hs4 4 0 191154276 chr4
chr - hs5 5 0 180915260 chr5
chr - hs6 6 0 171115067 chr6
...
You're also going to explore the randomdata Circos tool which can create random data for segments in a karyotype file. This is very useful for debugging and learning how to draw data because you know the statistical properties of the data.
>randomdata -karyotype karyotype.human.txt -ruleset default > random.txt
random.txt
hs1 0 9999999 0.0367
hs1 10000000 19999999 -0.0802
hs1 20000000 29999999 -1.3039
hs1 30000000 39999999 -0.4919
...
You're also going to see how to explore data on the command line with the histogram script that I have written for you.
> cat random.txt | ../../../scripts/histogram
-2.7940> 0 0.000
-2.7940 -2.4818 1 0.003 0.003
-2.4818 -2.1695 1 0.003 0.006
-2.1695 -1.8573 6 0.019 0.025 ***
-1.8573 -1.5451 9 0.028 0.053 ****
-1.5451 -1.2329 15 0.047 0.099 *******
-1.2329 -0.9206 22 0.068 0.168 ***********
-0.9206 -0.6084 25 0.078 0.245 ************
-0.6084 -0.2962 34 0.106 0.351 *****************
-0.2962 0.0161 41 0.127 0.478 ********************
0.0161 0.3283 45 0.140 0.618 **********************
0.3283 0.6405 50 0.155 0.773 *************************
0.6405 0.9528 24 0.075 0.848 ************
0.9528 1.2650 17 0.053 0.901 ********
1.2650 1.5772 18 0.056 0.957 *********
1.5772 1.8895 7 0.022 0.978 ***
1.8895 2.2017 2 0.006 0.984 *
2.2017 2.5139 4 0.012 0.997 **
2.5139 2.8261 0 0.000 0.997
2.8261 3.1384 0 0.000 0.997
3.1384 3.4506 1 0.003 1.000
3.4506< 0 0.000
n 322
average 0.02693
sd 0.94780
min -2.79400
max 3.45060
sum 8.67230
You'll use this script in other lectures to calculate statistical properties of data sets, such as the average gene size.
Color ramps
If there is enough time in the first session tomorrow, we'll look at creating color ramps.
These are color palettes like the Brewer palettes used to encode quantitative variables. You can play with ramps at
http://davidjohnstone.net/pages/lch-lab-colour-gradient-picker
to see how colors interpolate in different color spaces.
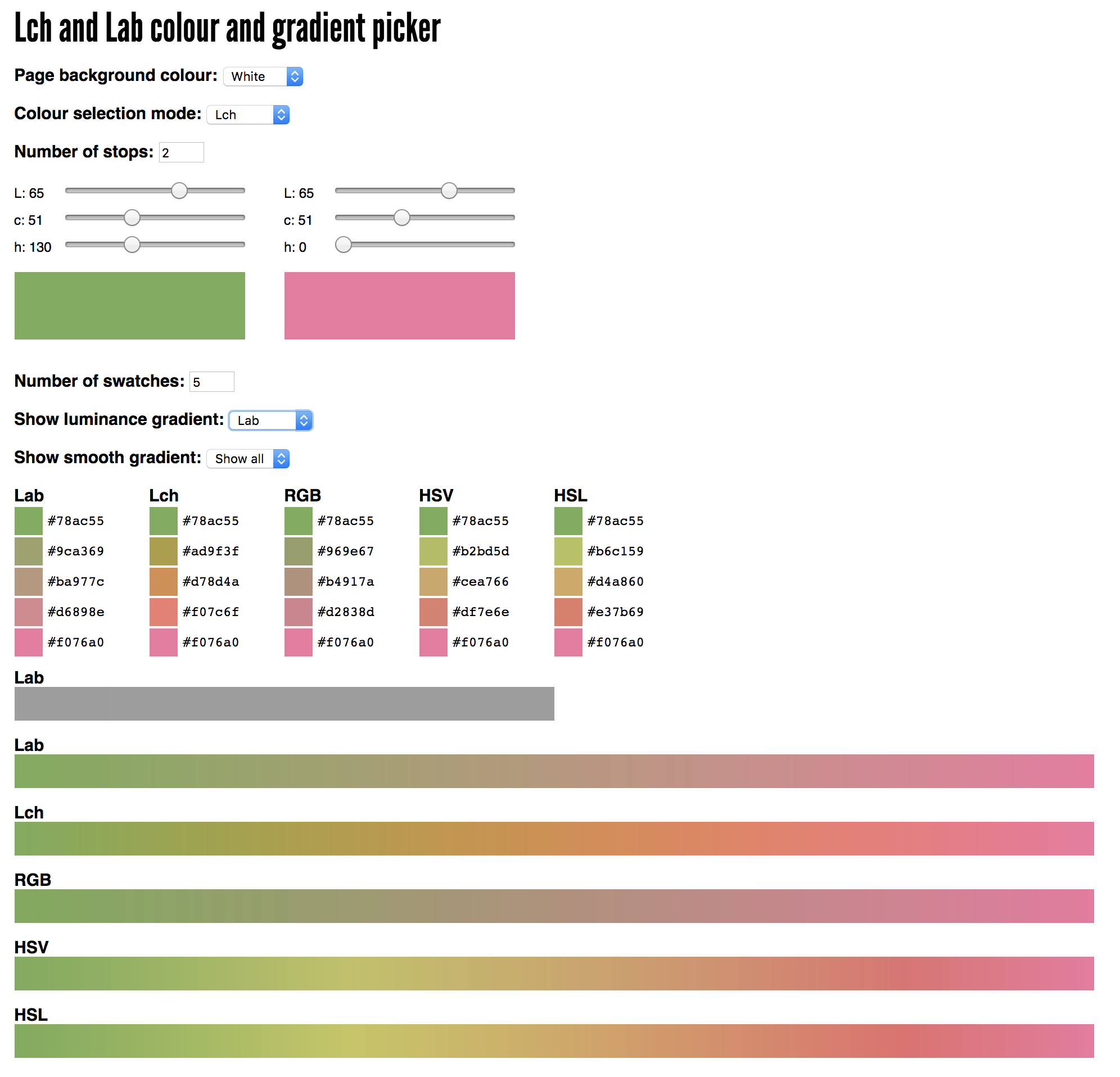
Because heatmap tracks take a list as a color (the values are mapped onto colors), this is a way for you to create your own.
Drawing human genes
In this lecture you'll get a chance to practise your command line parsing.
Using a list of 61,565 human genes (from UCSC genome browser) you will create a smaller list of about 3,500 genes.
hs19 58346805 58353499 8 name=A1BG
hs10 50799408 50885675 15 name=A1CF
hs12 9067707 9115962 36 name=A2M
hs12 8822471 8876783 36 name=A2ML1
hs1 33306765 33321098 5 name=A3GALT2
...
You will then use rules to select which gene families to draw. For example
<rule>
condition = var(name) =~ /^ZNF/
color = red
</rule>
<rule>
condition = 1
show = no
</rule>
would color red all genes whose name starts with ZNF and hide all others.

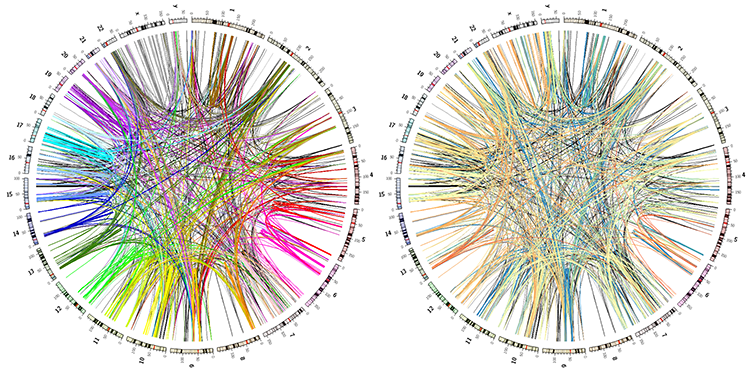
Drawing segmental duplications
You will practise more sophisticated command line parsing by filtering the file of human segmental duplications.
You'll create a file in the right format for Circos to draw links, but also with size ranks for each chromosome.
hs1 146541435 146905930 hs16 70811383 71168670 sizerank=1
hs1 148600078 148935345 hs1 119989247 120323081 sizerank=2
hs1 119989247 120323081 hs1 148600078 148935345 sizerank=3
...
hs2 110276210 110634615 hs2 109736854 110095177 sizerank=1
hs2 109736854 110095177 hs2 110276210 110634615 sizerank=2
hs2 94571013 94860516 hs9 65858856 66156287 sizerank=3
...
It's easy to sort the links by size across the entire genome
>awk '{print $3-$2,$0}' links.txt | sort -nr | cut -d " " -f 2- | awk '{print $0,"sizerank="NR}'
hsY 5464146 6234575 hsX 92352303 93120510 sizerank=1
hsX 92352303 93120510 hsY 5464146 6234575 sizerank=2
hsY 25545548 26311622 hsY 23358995 24124586 sizerank=3
hsY 23358995 24124586 hsY 25545548 26311622 sizerank=4
hsY 24894109 25541603 hsY 24128531 24775999 sizerank=5
hsY 24128531 24775999 hsY 24894109 25541603 sizerank=6
hsX 90276317 90909509 hsY 3853083 4483712 sizerank=7
hsY 3853083 4483712 hsX 90276317 90909509 sizerank=8
...
but it's trickier to sort them within each chromosome and assign a rank that is calculated to this within-chromosome sort.
You'll do this in bash by looping over all chromosomes
for chr in `cat track.segdup.all.txt | cut -d $'\t' -f 1 | sort -u` ; do
# 1. grep out the chromosome
# 2. sort by size
# 3. add a rank
# 4. append to temporary file
done
# 5. concatenate temporary files from each interation of the loop
You'll see that you can achieve a lot on the command line or with short bash scripts.
Having each link assigned a sizerank within a chromosome, you can then draw the 10 largest links for each chromosome by
<rule>
condition = var(sizerank) > 10
show = no
</rule>
which will hide all other links.
You'll also learn how to color data by chromosome. This is easy because there are colors named after chromosomes. This color scheme is the conventional color scheme used in the UCSC genome browser.

There is a color named chr1 as well as hs1 — both are the same color. Thus, any data point's chromosome name can be directly assigned to its color
<rule>
condition = 1
color = eval(lc var(chr1))
</rule>
or its luminance normalized equivalent. For example, to have all colors have L = 70,
<rule>
condition = 1
color = eval(sprintf("lum70%s",lc var(chr1)))
</rule>
In the images below, the image on the left has duplications colored by chromosome. The one on the right, by size.
Creating a sourvenir image
Finally, during the last session, you'll be able to compare your answers to the human gene and segmental duplication challenges.
You will also combine these two images into one.
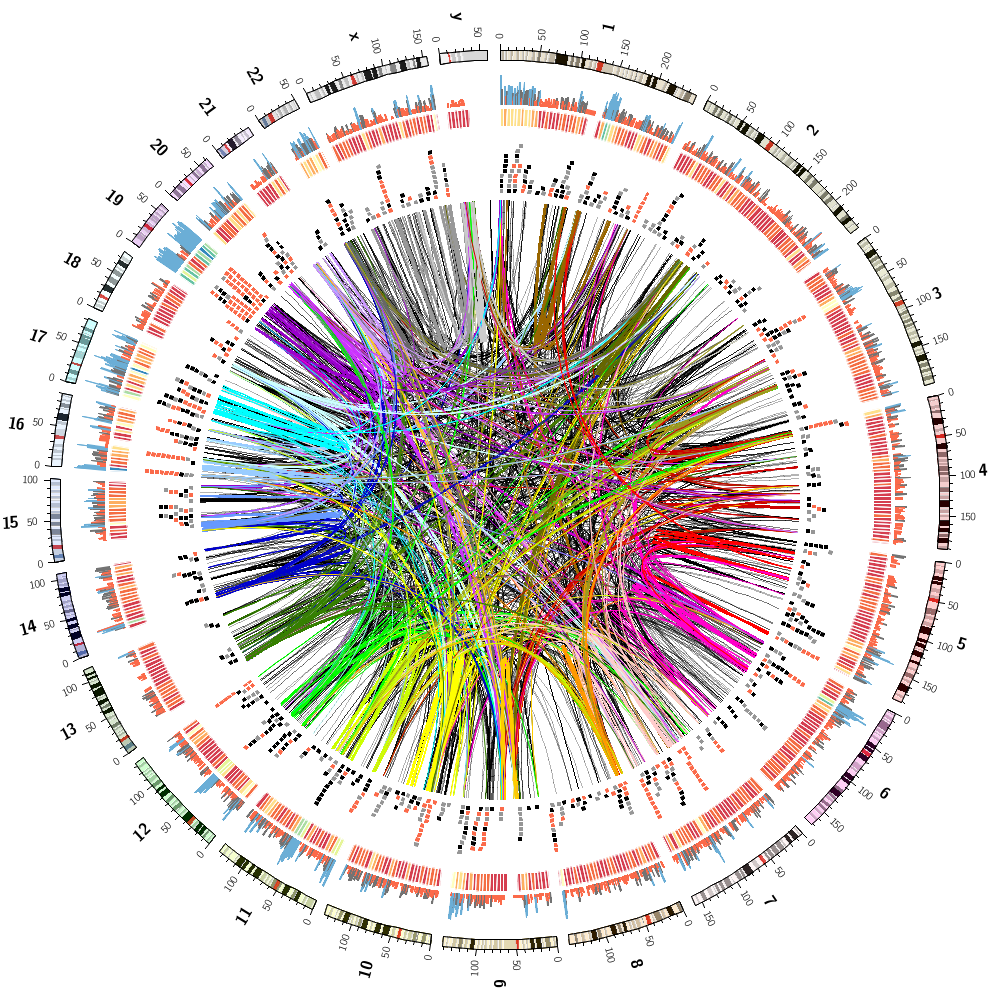
You'll then see how you can make a small adjustments in the configuration file to hide a random set of data! By changing the fraction of how much data is hidden, you can create progressively more complex images and then tile them.
For example, you can hide roughly half of the data by the rule
<rule>
condition = rand() < 0.5
show = no
</rule>
And you can define your own hiding fraction at the top of the configuration
hidefraction = 0.5
...
<plots>
<plot>
...
<rule>
condition = rand() < conf(hidefraction)
show = no
</rule>
</plot>
</plots>
You can then change the value of hidefraction in the configuration file, or force the change at the command line
>circos -param hidefraction=0.1 ...
This is very cool and lets you script Circos over loops of variables.
You'll also experiment with the -randomcolor flag which shuffles colors around in the image.
The examples below show Circos installation filesystem paths from an older course. Your setup will be different but probably not that different.
Verify Perl and Circos installation
Both Perl and Circos have been installed on your workstation.
To verify that both are in your PATH
> which perl
/BGA2017/perl-5.24.1/bin/perl
> which circos
/BGA2017/circos-0.69-5/bin/circos
If which does not return anything, you'll need to load the module
> module use /BGA2017/modulefiles
> module load circos
Alternatively, you would have received the directory where Circos was installed and if it's not in your PATH, you'll need to add it. For example, if Circos is installed in /opt/circos-0.69-6 then to add the Circos binary to your PATH at the command line
>export PATH=$PATH:/opt/circos-0.69-6/bin
You should also add this to your .bashrc file.
Download and Install Course Materials
Course materials can be downloaded from
http://mkweb.bcgsc.ca/pasteur/tunis.2018/circos.tunis.2018.v1.00.tgz
To install the materials, untar the archive, assuming you've downloaded it into your home directory.
# switch to your home directory
> cd ~
> tar xvfz ~/circos.tunis.2018.v1.00.tgz
Course Materials File Organization
The structure of the course materials is as follows
circos/
handouts/
sessions/
day.1/
lecture.1/
1/
2/
...
lecture.2/
...
day.2/
Material is organized by day and lecture (4 lectures per day). Each lecture has one or more independent sections. Here the term "lecture" covers both theoretical and practical lectures. For each day, lectures 2-4 are practical sessions in which you'll be working at your workstation.
The course material was originally conceived for 4 days. We have two days together, so I will be choosing lectures a la carte.
Using Course Files
During each lecture, you will be working entirely within the corresponding lecture directory.
For example, for Day 1 Lecture 2, you will be working from ~/circos-course/day.1/lecture.2
> cd circos/sessions/day.1/lecture.2
> ls
drwxr-xr-x 3 martink users 4096 Oct 15 10:32 1/
drwxr-xr-x 3 martink users 4096 Oct 15 10:32 2/
drwxr-xr-x 3 martink users 4096 Oct 15 10:32 3/
drwxr-xr-x 3 martink users 4096 Oct 15 10:32 4/
drwxr-xr-x 3 martink users 4096 Oct 16 19:44 5/
-rw-r--r-- 1 martink users 1509 Oct 16 19:40 README
On the course website you'll see the content of each of the files listed. For example, right now you're reading day.1/lecture.1/README
You'll start each lecture by navigating to its first directory
> cd circos/sessions/day.1/lecture.2/1
-rw-r--r-- 1 martink users 225865 Oct 16 19:42 circos.png
drwxr-xr-x 2 martink users 4096 Oct 16 19:41 etc/
and follow along on the webpage for that lecture. The page will show the contents of relevant files in this directory, such as one or more READMEs, Circos configuration and any scripts.
Once you are done with first part of the lecture, move to the next part
> cd ../2
Circos Configuration Files
Many lecture parts will have a etc/ directory with one or more Circos configuration files that are used to generate images for the lecture.
The main file configuration file will be etc/circos.conf file and other configuration files will be imported from that directory or others that have files shared between lecture. This configuration file will import contents from other files, such as (a) configuration files shared by other lessons in this session, (b) configuration files shared by all sessions in this course and (c) predefined configuration files in the Circos distribution that define default parameters.
Files are imported using the <<include>> directive. The included file is defined relative to the configuration file in which the <<include>> directive is found.
Lecture Structure
The structure of each lecture is the same. Files for lectures are independent — what you did in the previous lesson does not affect the configuration file of the next lesson.
Follow along on the webpage for the lecture and load the files being discussed in your text editor. Typically this will be etc/circos.conf but sometimes we'll be working with etc/ideogram.conf or etc/ticks.conf. It will be easier if you can have each file open in a separate editor buffer, or window.
If there is a README you can follow it along in your browser or text editor. It's up to you.
When you read files in your text editor, you're see some formatting codes such as code, file and link. These are used by the webpage for formatting. It should be pretty obvious when you see it.
Once you have the file loaded in your editor, you'll typically see some comments in the file that explain what is happening and what to do.
Frequently, you'll be asked to modify the file either by entering new content or by uncommenting lines that are commented out (lines prefixed with #).
If we don't have time to cover all the parts for a lecture, I encourage you to finish them on your own.
Creating Circos images
For lecture parts that have etc/circos.conf, you'll be creating Circos images.
To do this
> cd ~/circos/sessions/day.1/lecture.2/1
> circos
debuggroup summary 0.31s welcome to circos v0.69-7 2 Nov 2017 on Perl 5.010000
...
debuggroup output 11.27s generating output
debuggroup output 11.33s created PNG image ./circos.png (229 kb)
> ls
-rw-r--r-- 1 martink users 228897 Oct 19 13:28 circos.png
drwxr-xr-x 2 martink users 4096 Oct 16 19:41 etc/
Now look at the circos.png file in a file viewer.
*** IMPORTANT ***
You need to run Circos from the directory lecture.2/1 and not from lecture.2/1/etc or any other directory.
This is because there are relative path file definitions in the configuration files that refer to lecture directories on other days and these are defined relative to lecture.2/1.
If you get a file-not-found error, such as the one below, it's likely that you're running Circos from the wrong directory.
Error parsing the configuration file. You used an <<include FILE>> directive,
but the FILE could not be found. This FILE is interpreted relative to the
configuration file in which the <<include>> directive is used. Circos lookd
for the file in these directories
Circos Errors
If Circos cannot find the configuration file, you'll see this error
*** CIRCOS ERROR ***
CONFIGURATION FILE ERROR
Circos could not find the configuration file. To run Circos, you need to
specify this file using the -conf flag. The configuration file contains all
the parameters that define the image, including input files, image size,
formatting, etc.
If you do not use the -conf flag, Circos will attempt to look for a file
circos.conf in several reasonable places such as . etc/ ../etc
Common Errors
Watch out for these common errors when editing the configuration file. Circos can identify some errors and produce a detailed message.
Missing Configuration File
If you run Circos without specifying the configuration file with -conf and Circos cannot locate the file (it tries in etc/, ../etc and a few other places) you'll see this error
*** CIRCOS ERROR ***
CONFIGURATION FILE ERROR
Circos could not find the configuration file. To run Circos, you need to
specify this file using the -conf flag. The configuration file contains all
the parameters that define the image, including input files, image size,
formatting, etc.
If you do not use the -conf flag, Circos will attempt to look for a file
circos.conf in several reasonable places such as . etc/ ../etc
You can see where Circos tried to look by using -debug_group io
> circos --debug_group io
Unbalanced Configuration Blocks
Make sure that you do not forget to close a block.
<rules>
<rule>
</rule>
<rule>
</rules>
The missing </rule> for the second <rule> block will cause a parsing error. Don't forget closing </links>, </plots> and </rules> tags.
Redundant Parameter Definitions
In most cases you are not allowed to have multiple definitions of a parameter.
<plot>
..
color = black
..
color = red
..
</plot>
Circos will catch this and tell you what parameter is defined more than once.
CONFIGURATION FILE ERROR
Configuration parameter [color] in parent block [plot] has been
defined more than once in the block shown above, and has been interpreted as a
list. This is not allowed. Did you forget to comment out an old value of the
parameter?
Missing eval()
In a rule, if you want the parameter to be evaluated, don't forget eval().
<rule>
condition = 1
color = var(chr)
</rule>
This will set the color to var(chr) without evaluating it to the actual value of the chromosome. You want color = eval(var(chr))
When Circos does not recognize a color name it will default to black.
Debugging Circos
When things don't behave as you expect, you can use Circos' debugging facilities to narrow down the problem. Configuration Dump
Using the -cdump flag you can obtain the configuration file data structure. The output will reflect the hierarchical nature of the configuration file.
plots => {
color => 'black',
max => 1,
min => 0,
plot => [
{
file => '../data/both.cons.2e6.max.txt',
fill_color => 'spectral-5-div-3'
},
The output is large, and best combined with grep. For example, to search for all parameter that match cache,
> circos -cdump | grep cache
color_cache_file => 'circos.colorlist',
color_cache_static => 1,
Debug Groups
There is a large number of diagnostic reports that can be generated during image creation, named by the associated functionality. The reports can be accessed using -debug_group and combined as a list (e.g. io,conf,timer).
# reports about file location
>circos --debug_group io
# configuration file parsing and substitution
>circos --debug_group conf
# timings
>circos --debug_group timer
# all reports (long)
>circos --debug_group _all
For a list of report groups see
http://circos.ca/documentation/tutorials/configuration/debugging